
This sample Sickle-Cell Disease Research Paper is published for educational and informational purposes only. If you need help writing your assignment, please use our research paper writing service and buy a paper on any topic at affordable price. Also check our tips on how to write a research paper, see the lists of health research paper topics, and browse research paper examples.
Introduction
The sickling disorders are a group of inherited diseases of the hemoglobin (Hb) molecule within the erythrocyte/red blood cell (RBC) and comprise a substantial global public health issue. This group of diseases is found in high frequency in sub-Saharan Africa, the Mediterranean countries, the Middle East, and India but is also widely distributed throughout Europe and the United States, as well as Central and South America. A reduced oxygen tension allows the Hb S molecule to polymerize, causing distortion of the RBC into the characteristic half-moon shape. Sickled cells cause intermittent obstruction of the blood vessels (vasoocclusion) leading to ischemia (reduced blood flow) and infarction (tissue death) as well as anemia and other morbid complications.
Hemoglobin
Inside each RBC are millions of molecules of Hb. Four globin protein chains and four iron-binding protoporphyrin (heme) molecules make up each Hb molecule. The globin chains stabilize and solubilize the heme and facilitate oxygen uptake in the lungs and release in the tissues.
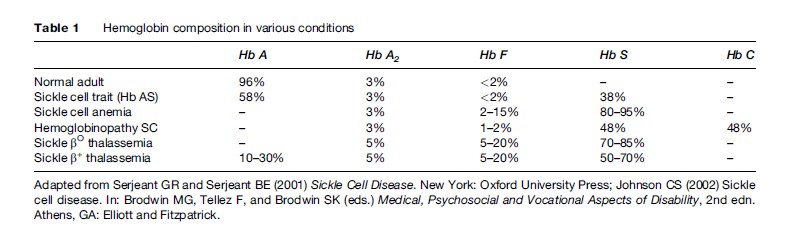
There are three types of normal hemoglobin, each made up of a different combination of the four normal globin chains (a, b, g, and d globins). Ordinarily, 96% of the normal adult hemoglobin composition is Hb A (two a and two b chains). Of the remaining hemoglobin, 3% is Hb A2 (two a and two d globins) and 1% is Hb F or fetal hemoglobin (two a and two g globins). The typical compositions of Hbs in normal and disease states are given in Table 1. Fetal hemoglobin is the primary type for the infant while in the uterus, as it is better able to take up oxygen from the relatively low levels present in the placenta. As a child ages, beta chain production gradually replaces gamma chains so that Hb A replaces Hb F by 6 months of age. At this time, the clinical features of a beta chain abnormality like Hb S become evident; signs of the disease such as hemolytic anemia are detectable, and symptoms of the disease may begin.
Genetics
There are two alpha chain genes located on chromosome 16, while single beta, gamma, and delta chain genes are on chromosome 11. Therefore, each chromosome in a pair controls half the total globin chain production. Each globin chain gene guides the RBC to make a specific globin chain; the globin chains match with a heme and then assemble into a complete Hb molecule. Hemoglobin disorders are of two main types: structural and synthetic. Structural hemoglobinopathies are due to a change in the amino acid sequence of either alpha or beta globin because of an alteration in the DNA sequence of the corresponding globin gene. Some of these genetic defects in the Hb molecule cause serious disease by causing anemia or by interfering with the oxygenation function of the molecule. On the other hand, many of these abnormal hemoglobins do not affect the way that Hb works and cause no symptoms at all. Synthesis defects are caused by a genetic change in the factors that control normal globin chain production, so that either alpha or beta globin is underproduced. This type of hemoglobinopathy produces a group of disorders known as the thalassemia syndromes; as in those with sickle cell trait, thalassemia is believed to provide partial protection from malaria and occurs in high prevalence in the Mediterranean area, the Middle East, and southeast Asia. More than 175 mutations producing beta thalassemia have been described. Alpha thalassemia is most often due to a deletion of one or more of the two alpha globin alleles on chromosome 16, but over 20 additional genetic defects producing alpha thalassemia have been described.
Nearly 700 hemoglobins that differ in structure or function from the normal molecule, Hb A, have been discovered. Basic information on these variants has been collected by Hardison et al. (1998). Sickle hemoglobin is due to a mutation in codon 6 of the beta globin gene, wherein an adenine to thymine substitution results in the substitution of valine for glutamic acid in the beta globin chain. This substitution allows the Hb S molecules to bind to one another when deoxygenated. Considerable scientific evidence indicates that the Hb S gene arose in several regions of Africa, the Mediterranean area, and some southeast Asian countries as partial protection against falciparum malaria.
Recent work has shown that some degree of the clinical heterogeneity is related to modifying genes that influence, positively or negatively, the clinical complications through modulation of Hb F expression, bilirubin catabolism, stroke, and iron loading, for example. The search for additional factors that contribute to the variability of disease expression depends upon demonstration of a cause-and-effect relationship, and the results of such studies are eagerly awaited.
The Sickle Cell Diseases
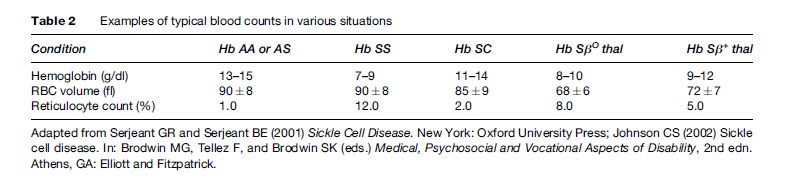
There are three common forms of sickle cell disease. Table 1 summarizes the hemoglobin composition in normals, in the common forms of sickle cell disease, and in the carrier state, sickle cell trait. Table 2 provides a typical picture of the blood counts in these conditions.
Sickle cell anemia (SCA) is the homozygous state, where the individual inherits the beta S gene from both parents, so that only Hb S is made. Hemoglobinopathy SC results from the inheritance of the gene for beta S from one parent and the gene for beta C from the other parent, so that both Hb S and C are produced in approximately equal amounts. The sickle thalassemias result from the inheritance of one gene for beta S and one for beta thalassemia. Consequently, the hemoglobin composition varies with the severity of the beta thalassemia gene defect, from zero production to near normal production (Table 1). Other hemoglobins may be co-inherited with Hb S but are less frequent; disease results when the second hemoglobin participates in the polymerization process. Hbs F and A inhibit polymerization and have an overall beneficial effect.
Pathophysiology: Sickle Cells, Anemia, And Obstruction Of Blood Flow
On deoxygenation, the presence of valine (Hb S) instead of glutamic acid allows adjacent Hb molecules to bind to each other. As additional binding occurs, polymerization of the Hb S molecules produces tubular fibers that distort the cell into the characteristic sickled shape. When the cell is reoxygenated, the Hb S fibers melt, and the cell returns to the biconcave (round) shape. Hb S polymerization occurs when the oxygen tension within the RBC is lowered but is accentuated when RBCs are exposed to acidosis (low pH), high temperature (fever), and increased osmolality (dehydration).
After several cycles of sickling and unsickling, the RBC becomes permanently damaged. A progressive loss of potassium (K+) and water results in a population of sickle RBCs of varying density. The denser RBCs increase the blood viscosity, with a reciprocal diminution in blood flow and increasing risk of vasoocclusion. These dense sickle RBCs are more rigid than the normal RBCs and are destroyed easily, shortening their life span to 15 days, rather than the normal 120 days. Bone marrow production of RBCs increases to keep up with the rate of destruction. Even at maximum output, the marrow is unable to manufacture enough cells to maintain a normal Hb level and chronic hemolytic anemia results. When RBCs are destroyed, the components of Hb are catabolized. The protoporphyrin in heme is broken down into bilirubin, which the liver excretes into the gallbladder as bile.
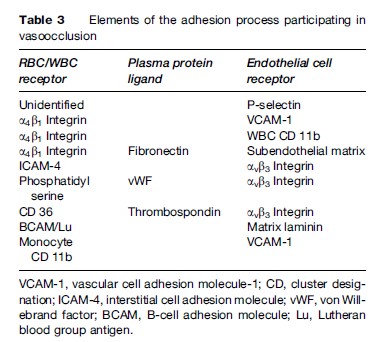
Vascular occlusion by sickle RBCs occurs at the postcapillary venule, leading to ischemia from lack of oxygen beyond the obstruction to blood flow, cell death (necrosis) in the poorly perfused area, and, ultimately, healing by fibrosis (scar tissue). Vasoocclusion is responsible for the signs and symptoms of this disease. The size of the obstructed vascular area and the amount and duration of the ischemia determine the degree of pain or organ damage. Although vascular occlusion can affect any of the body organs, certain vascular beds are particularly susceptible; these include the bone marrow, spleen, lung, kidney, and bones. Vasoocclusion in the sickling disorders is the result of a complex interaction involving the sickle RBC, the endothelial cells lining the blood vessels, leukocytes, and platelets, along with coagulation factors and other plasma proteins. Sickle RBC adherence to endothelium is the sine qua non of this interaction and is mediated by a number of cell surface receptors on the RBC and on the endothelial cell. Plasma proteins have a significant role as bridging molecules mediating adhesion pathways between RBCs and endothelial cells (Chiang and Frenette, 2005). A summary of these interactions is presented in Table 3.
Sickle Cell Anemia (SCA)
The most common form of sickle cell disease is SCA. Hemolytic anemia is present by the age of 6 months, although vascular occlusion symptoms may not appear until later. The anemia in SCA tends to be severe (Table 2). Much useful information on the clinical course of this disease has been the result of reports from the Cooperative Study of Sickle Cell Disease in the United States (Platt et al., 1991, 1994). Recurrent vasoocclusion leads to ischemic destruction of the spleen, which predisposes the patient to severe infections. Until recently, bacterial infections were the most common cause of death in children. Now, prophylactic treatment with antibiotics as reported by Gaston et al. (1986) has nearly eradicated this problem. Children with sickle cell anemia frequently have delayed growth and a delay in the onset of puberty. Delayed growth and sexual maturation appear to be related to nutritional factors, although the exact mechanism is still not clear.
There is wide variation in disease manifestations and severity (Platt et al., 1991). The long-term effects of recurrent vascular occlusion, ischemia, infarction, and fibrosis may cause one or more types of chronic organ damage, and the management of organ complications may become a major issue. The most common causes of death in these individuals are infections, stroke, lung disease, and chronic renal failure. The median survival determined by the Cooperative Study of Sickle Cell Disease is 42 years for males and 48 years for females (Platt et al., 1994), and appears to be improving in developed countries as the result of advanced technology and modern medical treatment. A low Hb F level, an elevated white blood cell (WBC) count, and severe anemia are predictors of premature mortality and reflect an underlying inflammatory state indicative of severe disease. Currently, hematopoietic stem cell transplantation is the only curative therapy for this disease. Its application is limited due to a lack of human leukocyte antigen-matched siblings without disease. However, over 175 transplants have been done with an overall survival over 90% and cure of the disease in over 80%. Additional trials of low-intensity preparative regimens or using unrelated donors are underway, attempting to find a way to expand the potential recipient population. Gene therapy remains an achievable goal, and much progress has been made in recent years. Investigators in the United States and in France are just beginning human trials of gene therapy (Bank et al., 2005).
Hemoglobinopathy SC
In hemoglobinopathy SC, Hb S and Hb C each make up half the hemoglobin composition (Table 1). Hemoglobinopathy SC is less common than SCA. By the time an infant is 6 months old, hemolytic anemia will be evident; however, vasoocclusive symptoms are often delayed until early adulthood. The anemia is less severe than that of SCA (Table 2). Crises and infections are less frequent than in SCA, although occasional patients have severe disease (Platt et al., 1991). Bone disease (aseptic necrosis) and retinal vessel involvement (sickle retinopathy) are more common than in SCA, whereas other types of organ damage are less common. The median survival of 60 years for males and 68 years for females is only slightly less than that in the U.S. African-American population
Sickle Thalassemias
Sickle thalassemia results from the inheritance of one beta S gene and a thalassemia defect on the other beta gene. In S beta thalassemia (βO ), the thalassemic globin gene prothe Hb S chromosome. In Sβ+ thalassemia, the thalassemic beta globin (b+) produces variable (low to near normal) amounts of protein, so that there is some Hb A present. The S-thalassemia’s are considerably less common than SCA or Hb SC in most populations. SβO thalassemia is similar to sickle cell anemia in its hematology, disease onset, course, and prognosis. Sβ+ thalassemia has minimal to moderate anemia, and its onset, course, and prognosis are similar to that of hemoglobinopathy SC (Platt et al., 1991).
Sickle Cell Trait
There is no disease associated with sickle cell trait. Those individuals with sickle cell trait have received one gene for beta S from one parent and one for beta A from the other parent. Although the RBCs of these individuals do not normally sickle in vivo, they can be induced to do so in the laboratory under extreme deoxygenation. Therefore, the RBC survival is normal, and there is no anemia. These individuals do not have the symptoms and signs of vasoocclusion, such as acute pain, frequent infections, or chronic organ damage, with two exceptions. Damage to the kidney may lead to hyposthenuria (inability to concentrate the urine), and splenic infarctions have been described in some. These two examples of end-organ damage are related to extreme environmental stress. Hyposthenuria is related to the high concentration of solute in the medulla of the kidney, which accentuates the sickling process locally and can damage the kidney concentrating mechanism. Splenic infarct is usually reported in persons exposed to high altitude, where exercise, hypoxia, dehydration, and acidosis may accentuate Hb S polymerization, leading to splenic infarct.

Specific Complications Of Sickle Cell Disease
Sickle cell crises, chronic hemolytic anemia, recurrent infections, acute and chronic organ damage, lower extremity ulceration, bone disease, and chronic nervous system damage are complications of sickle cell disease. Persons with sickle cell anemia are affected by these complications more often and with greater severity than those with either hemoglobinopathy SC or the sickle thalassemias. Within each genotype of the sickling diseases, there is a wide variation in the frequency of acute painful episodes, with a minority of patients having frequent episodes and the majority of patients having fewer (Platt et al., 1991).
Sickle Cell Crises
There are four types of sickle cell crises (Diggs, 1965). These are vasoocclusive, aplastic, splenic sequestration, and hyperhemolytic. The most common is the vasoocclusive (‘painful’) crisis. Vasoocclusive crisis has sudden onset, usually lasts 5 to 6 days, and may be localized in one area of the body or generalized. Recent evidence suggests that the pain is caused by bone marrow ischemia, which explains why there are so few changes detectable by physical examination and standard laboratory testing. For those severely affected by frequent episodes of pain, treatment with hydroxyurea has been recently shown by the Multi-Institutional Study of Hydroxyurea (Charache et al., 1995) to reduce the frequency of both pain events and acute chest syndrome by 45–50%. The suggestion that hydroxyurea reduces vasoocclusion has led to trials of this drug in young children to determine whether this drug will reduce the frequency or severity of end-organ damage (Ware et al., 2004).
Worsening anemia characterizes the three other kinds of crises. In the aplastic crisis, there is a temporary suppression of the RBC precursor cells in the marrow, usually by a viral infection. Splenic sequestration crisis is characterized by a bacterial infection that causes the spleen to enlarge rapidly, trapping RBCs and preventing them from re-entering the bloodstream. Hyperhemolytic crisis occurs due to infections, certain drugs, or toxins, and results in an acute increase in RBC destruction. In these types of crises, hemoglobin levels fall faster than the bone marrow can manufacture red blood cells, and the anemia becomes life-threatening. Aplastic, splenic sequestration, and hyperhemolytic crises are common only in SCA.
Hemolytic Anemia
Anemia is more severe in sickle cell anemia than in the other forms of the disease because in SCA, the RBCs are destroyed more rapidly (Table 2). Due to the need for increased RBC production, the bone marrow space enlarges to accommodate the increase in RBC production; consequently, the bones become remodeled and may produce bony deformities. The rapid RBC destruction produces large amounts of bilirubin from the protoporphyrin molecule in the heme; bilirubin breakdown products accumulate in the gall bladder, leading to stone formation as patients age. Anemia increases the workload of the heart in order to deliver adequate amounts of oxygen to the tissues. The increased workload on the heart causes easy fatigability and a decreased tolerance for physical activity.
Recurrent Infections
A major cause of morbidity and mortality in sickle cell disease, especially in children, has been bacterial infection. The frequency of these infections is several times that of the normal population. Although the exact reasons for this increased susceptibility to infection is poorly understood, one factor is the loss of splenic function caused by sickle cells. Septicemia, meningitis, osteomyelitis, pneumonia, and pyelonephritis are all more common in the sickle syndromes.
Kidney Disease
Hyposthenuria is nearly universal in the sickling diseases. Thus, replacement of urinary losses requires continuous intake of fluids in order to prevent RBC dehydration and its exacerbation of the sickling phenomenon. Ischemic damage leads to progressive renal failure and, eventually, the need for dialysis. Proteinuria is an important precursor manifestation of renal insufficiency, and its successful treatment appears to prolong the time to dialysis. Pyelonephritis is common in all forms of sickle cell disease.
Acute And Chronic Lung Disease
Acute pulmonary complications are second only to acute pain as a cause for hospitalization and are a predictor for early mortality. Despite aggressive diagnostic techniques, the exact etiology of most episodes is not elucidated, so that the term ‘acute chest syndrome’ is applied, acknowledging the inability to provide an exact diagnosis. Infection, especially viral illness, is common in childhood, while obstruction of pulmonary blood vessels by sickled cells, resulting in pulmonary infarction or fat or bone marrow embolization, seem more frequent in adults (Vichinsky et al., 1997, 2000). The most recent data on etiology are provided in Table 4. Although the signs and symptoms are similar to those of pneumonia, the clinical course in these patients is prolonged, suggesting a more complex pathophysiology involving vasoocclusion. Progressive pulmonary involvement can lead to respiratory insufficiency, for which exchange transfusion has been extremely beneficial, along with aggressive pulmonary support. Frequent pulmonary insults result in scarring of the lungs, which impairs gas exchange. Pulmonary insufficiency and pulmonary hypertension can occur. The reduced pulmonary function tends to exacerbate the sickling process because of low oxygen levels in the blood (Powars et al., 1988).

Eye Disease
Sickle cell retinopathy results from infarction of the retina and stimulation of new blood vessel growth into the vitreous cavity, similar to the neovascularization that occurs in diabetes mellitus. These abnormal vessels bleed or cause retinal detachment, resulting in loss of vision.
Leg Ulceration
Leg ulcers are common in those with sickle cell anemia, but less common in Hb SC and the sickle thalassemias. The etiology is felt to be poor blood flow coupled with venous insufficiency. A male preponderance has been reported in Africa and the United States, which is poorly understood (Serjeant et al., 2005).
Acute And Chronic Bone Disease
Acute arthritis may occur due to infarcts in the synovium, as well as bone infarcts. Since infarcted bone is an excellent culture medium for supporting bacterial growth, osteomyelitis (infection of a bone) or pyoarthrosis (infection of a joint) is a common complication of sickle cell disease. Vasoocclusion of the bones and joints may lead to osteonecrosis in the shoulders, hips, and vertebrae; the resultant collapse of the involved bone leads to degenerative arthritis, often requiring joint replacement.
Chronic Heart Disease
As a result of the anemia, cardiomegaly, left ventricular hypertrophy, and systolic flow murmurs are typical. The cardiac output is higher than normal, as related to the degree of anemia. It is unclear whether there is any direct cardiac dysfunction in this disease.
Pulmonary arterial hypertension has been recognized in 30–40% of adult patients with Hb SS and to a lesser degree in childhood and in the other sickle cell diseases. This complication is related to the hemolytic anemia, possibly through consumption of nitric oxide, a potent vasodilator, and is responsible for early mortality in those affected (Gladwin et al., 2004).
Central Nervous System Damage
Cerebral infarction is most common in childhood with a frequency of 10–13% at age 10 and a second peak in early adulthood. Detection of increased blood flow in the circle of Willis by transcranial Doppler ultrasonography identifies arterial narrowing and allows for prophylactic transfusion therapy with a significant reduction in both primary and secondary stroke prevention, as reported by Adams and colleagues (1998). Intracranial hemorrhage occurs in young adults with a high immediate mortality. In addition, there is increasing recognition of ‘silent stroke’ affecting executive functions (decision making, educational attainment, etc.) in these patients.
Liver Disease
Transfusion therapy in used more frequently for the acute and chronic complications of sickling. Consequently, transfusion-transmitted hepatitis B and C are common manifestations in this disease. Iron overload with hepatic cirrhosis occurs unless chelation therapy is effective. Iron accumulation in the heart muscle may produce left ventricular dysfunction. Sequestration of RBCs in the liver can cause severe anemia, as in splenic sequestration, accompanied by severe hepatic dysfunction.
Pregnancy
Current pregnancy outcomes are substantially improved over pre-1975 data (Hassell, 2005); maternal mortality, fetal mortality, and miscarriage rates in developed countries are considerably lower than in older studies. A decreased uteroplacental blood flow correlates with low birth weight, but uteroplacental blood flow is not improved following transfusion and reduction of sickle cells, suggesting a vascular cause for the abnormal flow. Multiple studies now demonstrate that the degree of anemia does not correlate with fetal outcome. Importantly, a randomized trial of prophylactic transfusion during pregnancy failed to show any benefit in obstetric complications nor an improvement in fetal birth weight or reduction in intrauterine growth retardation rates (Koshy et al., 1988). There was a reduction in the number of acute painful events but not in the complications of sickle cell disease (e.g., acute chest syndrome).
Priapism
Priapism is a persistent, unwanted erection lasting more than 30 min. The mechanism is sickling and sludging of blood in the corpora cavernosa, with failure of detumescence. It typically occurs during sleep, when relative nocturnal acidosis and dehydration favor RBC sickling. From 30–45% of males report instances of priapism, either stuttering or prolonged. A single episode is more common in childhood. Impotence is common after prolonged episodes.
Summary
The sickling disorders have multisystem effects that require ongoing surveillance for detection of evolving end-organ dysfunction and institution of appropriate therapy. Despite recent advances in diagnosis and therapy, these diseases continue to consume considerable health-care resources and have a generally poor quality of life and premature mortality. Transfusion and hydroxyurea are the mainstays of current treatment; additional therapies are in development and await demonstration of efficacy in the clinic. Stem cell transplantation is available to a few, but gene therapy remains the hope for the future.
Bibliography:
- Adams RJ, McKie VC, Hsu L, et al. (1998) Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New England Journal of Medicine 339: 5–11.
- Bank A, Dorazio R, and Leboulch P (2005) A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Annals of the New York Academy of Sciences 1054: 308–316.
- Charache S, Terrin ML, Moore RD, et al. (1995) Effects of hydroxyurea on the frequency of painful crises in sickle cell anemia. New England Journal of Medicine 332: 1317–1322.
- Chiang EY and Frenette PS (2005) Sickle cell vasoocclusion. Hematology/Oncology Clinics of North America 19: 771–784.
- Diggs LW (1965) Sickle cell crises. American Journal of Clinical Pathology 44: 1–19.
- Gaston MH, Verter JI, Woods G, et al. (1986) Prophylaxis with oral penicillin in children with sickle cell anemia. New England Journal of Medicine 314: 1593–1599.
- Gladwin MT, Sachdev V, Jison ML, et al. (2004) Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. New England Journal of Medicine 350: 886–895.
- Hardison R, Chui DHK, Riemer C, et al. (1998) Access to A Syllabus of Human Hemoglobin Variants (1996) via the World Wide Web. Hemoglobin 22: 113–127.
- Hassell K (2005) Pregnancy and sickle cell disease. Hematology/ Oncology Clinics of North America 19: 903–916.
- Johnson CS (2002) Sickle cell disease. In: Brodwin MG, Tellez F, and Brodwin SK (eds.) Medical, Psychological and Vocational Aspects of Diability, 2nd edn. Athens, GA: Elliot and Fitzpatrick.
- Koshy M, Burd L, Wallace D, et al. (1988) Prophylactic red-cell transfusions in pregnant patients with sickle cell disease. A randomized cooperative study. New England Journal of Medicine 319: 1447–1452.
- Platt OS, Brambilla DJ, Rosse WF, et al. (1994) Mortality in sickle cell disease: Life expectancy and risk factors for early death. New England Journal of Medicine 330: 1639–1644.
- Platt OS, Thorington BD, Brambilla DJ, et al. (1991) Pain in sickle cell disease. New England Journal of Medicine 325: 11–16.
- Powars D, Weidman JA, Odom T, Niland J, and Johnson C (1988) Sickle cell chronic lung disease: Prior morbidity and the risk of pulmonary failure. Medicine (Baltimore) 67: 66–76.
- Segeant GR and Segeant BE (2001) Sickle Cell Disease. New York: Oxford University Press.
- Serjeant GR, Serjeant BE, Mohan JS, and Clare A (2005) Leg ulceration in sickle cell disease: Medieval medicine in a modern world. Hematology/Oncology Clinics of North America 19: 943–956.
- Vichinsky EP, Neumayr LD, Earles AN, et al. (2000) Causes and outcomes of the acute chest syndrome in sickle cell disease: National acute chest syndrome study. New England Journal of Medicine 342: 1855–1865.
- Vichinsky EP, Styles LA, Colangelo H, et al. (1997) Acute chest syndrome in sickle cell disease: Clinical presentation and course; Cooperative study of sickle cell disease. Blood 89: 1787–1792.
- Ware RE, Zimmerman SA, Sylvestre PB, et al. (2004) Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. Journal of Pediatrics 145: 346–352.
- Barton JC, Lee PL, Bertoli LF, and Beutler E (2005) Iron overload in an African American woman with SS hemoglobinopathy and a promoter mutation in the X-linked erythroid-specific 5-aminolevulinate synthase (ALAS2) gene. Blood Cells Molecules and Diseases 34: 226–228.
- Chaar V, Keclard L, Diara JP, et al. (2005) Association of UGT1A1 polymorphism with prevalence and age at onset of cholelithiasis in sickle cell anemia. Haematologica 90: 188–199.
- Hoppe CC and Walters MC (2001) Bone marrow transplantation in sickle cell anemia. Current Opinion in Oncology 13: 85–90.
- Johnson CS (2005) Pulmonary complications of sickle cell disease: The acute chest syndrome. In: Sharma OP (ed.) Lung Biology in Health and Disease, pp. 475–505. New York: Taylor and Francis.
- Ofori-Acquah SF, Lalloz MR, Serjeant G, and Layton DM (2004) Dominant influence of gamma-globin promoter polymorphisms on fetal haemoglobin expression in sickle cell disease. Cellular and Molecular Biology 50: 35–42.
- Platt OS, Rosenstock W, and Espeland M (1984) Influence of sickle cell hemoglobinopathies on growth and development. New England Journal of Medicine 311: 7–12.
- Puthenveetil G and Malik P (2004) Gene therapy for hemoglobinopathies: Are we there yet? Current Hematology Reports 3: 298–305.
- Serjeant GR (1992) Sickle Cell Disease, 2nd edn. Oxford, UK: Oxford University Press.
- Steinberg MH, Forget BG, Higgs DR and Nagel RL (eds.) (2001) Disorders of Hemoglobin. Cambridge, UK: Cambridge University Press.
- Steinberg MH, Barton F, Castro O, et al. (2003) Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. Journal of the American Medical Association 289: 1645–1651.
- Stuart J and Johnson CS (1987) Rheology of the sickle cell disorders. Balliere’s Clinical Hematology 1: 747–775.
- Taylor JG, Tang DC, Savage SA, et al. (2002) Variants in the VCAM-1 gene and risk for symptomatic stroke in sickle cell disease. Blood 100: 4303–4309.
See also:
Free research papers are not written to satisfy your specific instructions. You can use our professional writing services to buy a custom research paper on any topic and get your high quality paper at affordable price.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality



