
This sample Metabolic Myopathies Research Paper is published for educational and informational purposes only. If you need help writing your assignment, please use our research paper writing service and buy a paper on any topic at affordable price. Also check our tips on how to write a research paper, see the lists of health research paper topics, and browse research paper examples.
Background
The metabolic myopathies represent a group of muscle disorders characterized by impairments in intermediary metabolism. The three main categories of disease include the mitochondrial myopathies, fatty acid oxidation defects (FAODs), and glycogen storage diseases (GSDs).
Myoadenylate deaminase deficiency (AMPD1) has been considered by some to be a metabolic myopathy; however, its prevalence of almost 2% in the general population, and the documentation of completely asymptomatic individuals, renders its pathogenicity in question. Although there are a number of medications that can impair muscle metabolism (e.g., statins), these are generally grouped within the category of toxic myopathies and will not be considered further in this research paper. The focus of this research paper will be on the three major categories of inborn errors of metabolism as described above. It is important to recognize these conditions, because interventional strategies may be effective in preventing rhabdomyolysis and the severe consequence of potential renal failure. Furthermore, in conditions such as mitochondrial myopathies there may be multisystem involvement with very severe consequences, and genetic counseling may be appropriate. Finally, multisystem disorders such as the mitochondrial myopathies may be mislabeled as multiple sclerosis (MS), nonspecific encephalopathy, or vasculitis, and accurate diagnosis can avoid inappropriate therapeutic interventions such as b-interferon for MS or cyclophosphamide for vasculitis. A list of the more common metabolic myopathies is found in Table 1.
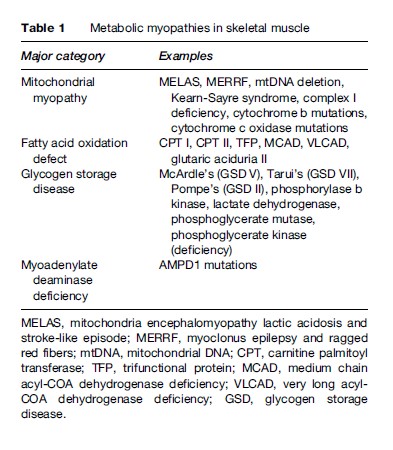
Although some consider the metabolic myopathies to be rare entities, it has been estimated that mitochondrial cytopathies affect about 1 in 8000 in the population. Although the FAODs and GSDs are comparatively less common (McArdle’s disease = – 1/100 000; carnitine palmitoyl transferase (CPT) deficiency 1/300 000, respectively), they do represent one of the more common causes of exercise-induced rhabdomyolysis, and prompt recognition by the treating physician is important.
The purpose of this research paper is to present a brief overview of the more common metabolic myopathies to provide the clinician with the tools to recognize symptoms, organize appropriate diagnostic testing, and be familiar with the available treatment options. Given space constraints, the reader will be referred to comprehensive and contemporary reviews. Other reviews of metabolic myopathies in general can be found in the references by Tarnopolsky (2006) and Tein (1996).
General Features Of The Disorders And Inheritance
Mitochondrial Disorders
Mitochondrial disorders represent a diverse group of conditions with a primary defect in electron transport chain function. Although other conditions, such as amino acid and fatty acid oxidation defects, also occur in the mitochondria, they are not traditionally considered part of the mitochondrial disorders. Many of the mitochondrial myopathies were originally characterized by acronyms based upon the phenotypic presentation. For example, MELAS refers to Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes. The era of mitochondrial medicine expanded rapidly after the identification of a point mutation responsible for Leber’s hereditary optic neuropathy (LHON, G11778A), chronic progressive external ophthalmoplegia (CPEO, mitochondrial DNA deletions) and MELAS (A3243G). Since that time there has been a massive proliferation of the number of point mutations in the mitochondrial genome ascribed to phenotypic characteristics.
In addition to the vast array of mutations found within the maternally inherited mitochondrial DNA (mtDNA), there is an increasing recognition of the mitochondrial disorders arising from mutations within the nuclear genome and following Mendelian genetic inheritance patterns. For example, the mutations responsible for a number of autosomal recessive conditions have been found, including myo-neuro-gastrointestinal encephalomyopathy (MNGIE, thymidine phosphorylase), complex I (NDUF) and IV (SURF-1) Leigh’s disease, mtDNA depletion (dGuOK, TK) and some forms of mtDNA deletion syndromes (polymerase gamma, twinkle, ANT). Autosomal dominant inheritance patterns have also been seen in rare cases of mtDNA deletion syndromes.
Irrespective of the mutation, many of the cellular consequences of mitochondrial dysfunction can be linked to a decrease in aerobic energy production and/or an increased production of free radicals. Given that the mitochondrion is the final common pathway for the oxidative decarboxylation of fats, proteins, and carbohydrates, it is understandable that mitochondrial dysfunction can impair cellular energy metabolism, which can be particularly apparent during periods of superimposed metabolic stress (exercise, infection, prolonged fasting). Given the ubiquitous presence of mitochondria in all tissues except red blood cells, there is often widespread tissue involvement (mitochondrial cytopathies); however, most patients will have varying degrees of muscle symptoms (decreased endurance or weakness), and these forms can be termed ‘mitochondrial myopathies.’ Recent reviews of the mitochondrial cytopathies can be found in Tarnopolsky and Raha (2005) and Dimauro et al. (2006).
Fatty Acid Oxidation Defects
FAODs ultimately impair b-oxidation of lipid within the mitochondrial matrix. The main defects that have been identified include transport of long-chain fat across the mitochondrial membrane (i.e., CPT deficiency); transport of carnitine into the cell (i.e., carnitine transporter deficiency); and the majority of defects attributed to mutations in b-oxidation directly (i.e., long-chain acyl-CoA dehydrogenase (LCAD), medium-chain acyl-CoA dehydrogenase (MCAD), and trifunctional protein (TFP) deficiencies). These disorders are inherited with an autosomal recessive inheritance pattern. The more severe variants present in infancy or childhood with a primary liver or encephalopathic picture, while the adult-onset forms are predominantly myopathic. The main FAODs presenting in adulthood include CPT II, TFP, and very-long-chain acyl-CoA dehydrogenase deficiencies. Further reading in this area can be found in the reviews by Tein (1996) and Vockley et al. (2002).
Glycogen Storage Disease
GSD refers to a group of disorders characterized by genetic mutations in glycogen synthesis, glycogenolysis, or glycolysis. The pathology results from an inability to break down glycogen to maintain plasma glucose concentration (e.g., hepatic forms such as hepatic phosphorylase deficiency or glucose-6-phosphatase deficiency), abnormal tissue storage and cirrhosis (e.g., branching enzyme deficiency), or the myopathic forms that inhibit muscle glycogenolysis or glycolysis (e.g., McArdle’s disease, Tarui’s disease, etc.). The hepatic forms usually result in hypoglycemia as the main clinical manifestation, with type IV also leading to cirrhosis due to the accumulation of abnormal nonbranched glycogen molecules. The myopathic forms usually result in muscle cramping and premature fatigue particularly during high-intensity exercise when there is the obligatory use of anaerobic pathways. Some of the myopathic forms of GSD result in fixed muscle weakness such as Pompe’s disease (GSD II), whereas others such as McArdle’s disease (GSD V) may develop a more indolent proximal myopathy later in life. With the exception of phosphorylase b kinase deficiency and phosphoglycerate kinase deficiency, which are X-linked recessive conditions, the remainder of the GSDs are autosomal recessive in their inheritance pattern.
The more common myopathic forms of GSD, in approximate order of frequency, include phosphorylase deficiency (McArdle’s disease, GSD V), acid maltase deficiency (Pompe’s disease, GSD II), phosphorylase b kinase deficiency (GSD VIII), and phosphofructokinase (PFK) deficiency (Tarui’s disease, GSD VII). Less common myopathic forms include debranching enzyme (GSD III), phosphoglycerate kinase (GSD IX), phosphoglycerate mutase (GSD X), and lactate dehydrogenase (GSD XI) deficiencies.
Clinical Presentation
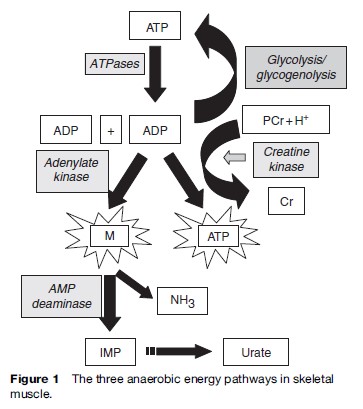
It is important to note that many of the metabolic myopathies present with symptoms during exercise. During higher intensity exercise such as sprinting, or at the onset of aerobic exercise, there is predominantly an anaerobic component, with the adenylate kinase/myoadenylate deaminase pathway being quantitatively the least important, followed by the CK-mediated pathway of phosphocreatine hydrolysis, and anaerobic glycolysis and glycogenolysis (Figure 1).
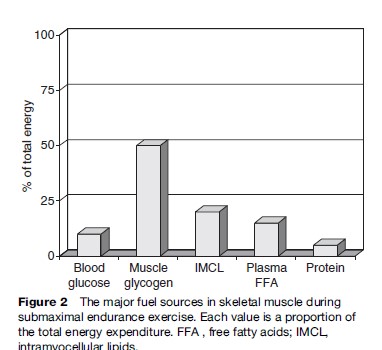
With endurance-type activity, the proportion of oxidized fuels changes as a function of exercise intensity, training status, and gender; however, at moderate-intensity exercise (less than 50% of maximal aerobic capacity (VO2max)), the main sources of fuels are plasma free fatty acids and blood-borne glucose. As the intensity of exercise increases there is proportionately greater utilization of intramuscular glycogen and intramyocellular lipids, with almost exclusive utilization of intramuscular glycogen at intensities closer to a 10-km or marathon race pace. The mitochondria are obligatory for the oxidative metabolism of carbohydrate fat and protein, thus representing the final common pathway of aerobic energy transduction. The main fuel sources during endurance activity at approximately 65% of the VO2max are presented in Figure 2.
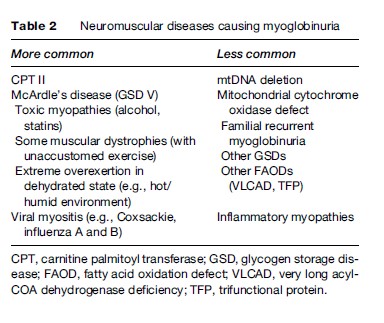
Under normal circumstances, the ATP content of skeletal muscle is tightly regulated and does not decrease; however, with the metabolic myopathies it is possible to reach a state of metabolic crisis and induce rhabdomyolysis, which can lead to myoglobinuria and subsequent renal failure. A list of some of the causes of myoglobinuria is found in Table 2. A list of the common symtoms of the metabolic myopathies is presented in Table 3.
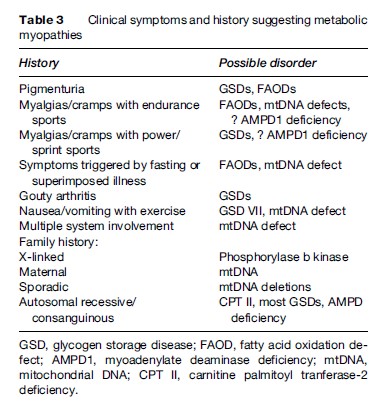
Mitochondrial Disorders
There is extreme phenotypic and genotypic heterogeneity in the mitochondrial cytopathies. For example, patients with the MELAS A3243G gene mutation can present with later adult-onset deafness and diabetes, or in infancy with fatal encephalomyopathy with seizures and stroke-like episodes. Conversely, patients with a wide array of specific point mutations, mtDNA deletions, and nuclear defects can present with encephalopathy, muscle fatigue or ptosis and hearing loss. Due to the impairment in aerobic energy transduction, most patients have very low maximal oxygen uptake (VO2max), which renders daily activities much more stressful and taxing and patients often present with exercise intolerance. Although the exercise intolerance is often overshadowed by more significant neurological symptoms, most adult patients will have quite severe exercise intolerance, often dating back to childhood. These individuals were often labeled as being ‘the worst athlete in the school’ and frequently avoided physical activity. Unlike the FAODs and GSDs, it is rare for patients to have rhabdomyolysis with resultant myoglobinuria; however, this can be seen in cytochrome b, cytochrome c oxidase (COX), and MELAS A3260G mutations.
Figure 2 The major fuel sources in skeletal muscle during submaximal endurance exercise. Each value is a proportion of the total energy expenditure. FFA , free fatty acids; IMCL, intramyocellular lipids. GSD, glycogen storage disease; FAOD, fatty acid oxidation defect; AMPD1, myoadenylate deaminase deficiency; mtDNA, mitochondrial DNA; CPT II, carnitine palmitoyl tranferase-2 deficiency.
In addition to exercise intolerance, patients with mitochondrial myopathies frequently have headache, nausea, and vomiting induced by exercise and can become frankly encephalopathic. We have seen one pedigree with the MELAS A3260G gene mutation where exercise-induced deafness was a characteristic feature. The symptoms of exercise intolerance are much worse when individuals have a superimposed infection or are in the fasted state, which are conditions where a greater reliance on mitochondrial energy function is required.
In addition to exercise intolerance and the extensive list of other clinical features that can accompany these disorders (ptosis, ophthalmoplegia, hearing loss, strokelike episodes, migraine, headaches, cardiomyopathy,ataxia, encephalopathy, etc.), another muscle-related symptom can be fixed weakness. We have had several pedigrees with the MELAS A3243G gene mutation where fixed proximal weakness to the point of respiratory failure was a presenting feature. In most patients, however, strength is reasonably well preserved, and the factors predisposing some patients to severe weakness are currently unclear.
Fatty Acid Oxidation Defects
Many patients with myopathic FAODs are asymptomatic without the superimposition of a metabolic stress. In children, the two main metabolic stressors are superimposed illnesses, such as viral illness, or prolonged fasting, nausea, and vomiting with decreased fluid and caloric intake, where significant weakness, lethargy, and even encephalopathy can occur. The adult myopathic forms of FAOD usually present during endurance type activity, particularly if it is prolonged, performed in the fasted state, or with a superimposed viral illness. These individuals can often perform high-intensity activity for short periods of time without difficulty; however, when the utilization of lipid becomes more important, such as during longer-term endurance activity, they often experience cramps, muscle discomfort, and inability to continue the activity. There is often pigmenturia later in the day or the next day following such an activity, and the muscles remain very sore for several days. In some cases severe myalgias with rhabdomyolysis and renal failure can be the presenting symptom of FAOD.

Glycogen Storage Disease
As mentioned previously, the infantile and childhood forms of GSD often present with hypoglycemia and an encephalopathic picture. McArdle’s disease (GSD V) is exclusively a myopathic condition with muscle cramps and pigmenturia induced by higher-intensity exercise, such as climbing stairs or sprinting for a bus. Other disorders, such as Tarui’s disease and phosphorylase b kinase deficiency (GSD VIII), are usually predominantly myopathic; whereas all of the other myopathic forms can have more severe pediatric onset, with hepatomegaly and more severe progressive proximal myopathy in addition to exercise intolerance. Patients with Pompe’s disease (GSD II) have predominantly fixed proximal weakness and respiratory insufficiency rather than exercise-induced discomfort. Patients with McArdle’s disease and Tarui’s disease have a compensatory increase in flux through the adenylate kinase/myoadenylate deaminase pathway, with a subsequent increase in uric acid, and can present with myogenic hyperuricemia and gout.
Diagnostic Testing
The main diagnostic tests for the metabolic myopathies include blood tests (serum CK activity, lactate, acylcarnitine), urine testing (organic acids, myoglobin), muscle biopsy (light and electron microscopy, enzymology), exercise testing, genetic testing, and magnetic resonance spectroscopy. These should be conducted at a center with experience in these disorders and the ability to collect and process samples appropriately. For example, delays in freezing the muscle biopsy, delays in serum lactate determination, and timing of sample acquisition are only a few examples of situations in which false-positive or -negative tests can occur. A summary of the common diagnostic tests can be found in Table 4.

Mitochondrial Disorders
The serum lactate determination is one of the more important blood tests. This indicator is elevated in approximately 70% of patients. It is important that the blood be collected on ice and promptly analyzed. An elevated lactate has high specificity (>0.9) but lower sensitivity ( 0.7) for mitochondrial cytopathies. Falsepositive tests can occur in diabetics and in patients who ate a high-carbohydrate meal within 2 h of collection. Patients with MELAS may have elevated plasma glucose level or impaired glucose tolerance given their propensity towards insulin resistance in type 2 diabetes. Serum CK activity may be elevated but is rarely more than five times the upper limit of normal. Urine testing includes organic acid assessment with elevations noted in ethylmalonic acid, 3-methyl glutaconic acid, and fumarate.
The muscle biopsy can show ragged red fibers (subsarcolemmal accumulation of mitochondria on modified Gomori trichrome staining), COX-negative fibers, and accumulation of neutral lipids. At the electron microscopic level, pleomorphic mitochondria often containing paracrystalline inclusions are a feature of mitochondrial dysfunction. Although some of the mitochondrial myopathies will show defects in specific enzyme activity in fibroblasts and platelets, the vast majority are detectable only with biochemical assays of electron transport chain enzyme activity in skeletal muscle homogenates or isolated mitochondria. A selected defect in complex IV activity would prompt a search for mutations associated with complex IV assembly (SCO2) or mutations in the mitochondrial (COI, II, III) or nuclear-encoded (COIV, COVb) sub-units. Conversely, multiple enzymatic defects would prompt a search for transfer RNA or mtDNA mutations.
Specific mutation analysis can be completed if a given gene mutation is strongly suspected from the history or if a mutation is known in a family. Mutation analysis can be performed using polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP), allele specific oligonucleotide, and a host of other tests to identify the point mutation. Often, a test panel is completed as a function of the clinical phenotype. For example, a patient with stroke-like episodes, seizures, and muscle fatigability would probably start with a screen for MELAS A3243G, T3271C. A patient with CPEO would prompt a search for mtDNA deletions by Southern blot or long-range PCR. If a mutation is not identified, mtDNA sequencing can be completed in approximately one day of the entire genome (16 569 base pairs).
Exercise testing in mitochondrial myopathies usually demonstrates a markedly reduced VO2max or an abnormally high respiratory exchange ratio (indicative of early lactate production) with cycle ergometry testing (for a review, see Tarnopolsky, 2004). A variety of testing modalities have been employed with forearm exercise testing, including near infrared spectroscopy or direct venous blood gas measurements, to demonstrate the lack of deoxygenation due to decreased oxygen consumption by defective mitochondria. Phosphorus (31P) magnetic resonance spectroscopy has also been used to demonstrate an increased reliance on phosphocreatine hydrolysis or a delay in the postexercise phosphocreatine recovery. A review of the diagnostic criteria for the mitochondrial cytopathies can be found in Naviaux (2004).
Fatty Acid Oxidation Defects
Blood tests including CK, lactate, and glucose can be completely normal in asymptomatic individuals with FAODs. During an acute metabolic crisis, there is often hypoketotic hypoglycemia and an increase in serum CK activity. Other findings such as hyperkalemia can be associated with an acute episode of rhabdomyolysis. If the rhabdomyolysis induces renal failure, a delayed increase in urea and creatinine and other markers such as metabolic acidosis and hyperkalemia can accompany the acute renal failure.
More definite blood testing can be completed using liquid chromatography–tandem mass spectroscopy (LC/ MS/MS) characterizing the acyl-carnitine profile. Occasionally, in the non-stressed situation, but more often during an acute event, the acyl-carnitine signature can yield the definitive diagnosis. A reduction in serum total and free carnitine can also be documented in many cases.
Urine testing during an acute myopathic event may show myoglobin, in which case admission and standard rhabdomyolysis protocols must be employed. During this time, dicarboxylic acids can be identified in the urine using gas chromatography/mass spectroscopy (GC/MS). Enzymatic testing in fibroblasts is usually much more informative than with the mitochondrial cytopathies and specific enzyme pathways (CPT II) or functional assays with mediumor long-chain substrates can be used to help identify a biochemical defect. The molecular defects can be screened using standard procedures once either an acyl-carnitine profile or biochemical tests suggest a specific mutation. In some cases, such as a classic presentation of CPT II deficiency, it may be appropriate to screen by checking for the most common mutation (ser113leu) through a variety of genetic methods as mentioned previously.
Exercise testing is often normal, although the VO2max can be somewhat diminished; however, this is neither sensitive nor specific. Occasionally, a high respiratory exchange ratio during low-intensity exercise may indicate the increased reliance upon carbohydrates, although this finding cannot rule in or rule out a FAOD. A muscle biopsy may show an increase in neutral lipids but may also be completely normal, with no other structural alterations. Reviews of the FAODs can be found in Tein (1996).
Glycogen Storage Disease
The serum CK activity is invariably elevated in patients with McArdle’s disease at all times and may be variably elevated in the other GSDs. During an acute metabolic crisis the CK activity is invariably elevated to values that can exceed 100 000 U/L. The elevation of CK activity and the resultant risk for renal abnormalities are no different from those seen for FAODs, and these patterns alone cannot be used to determine the etiology of the rhabdomyolysis during an acute event. Serum uric acid may be elevated due to myogenic hyperuricemia, and this can trigger gouty arthritis. A unique feature of PFK deficiency is the presence of hemolytic anemia. Urine testing is totally normal except during acute rhabdomyolysis, where the expected myoglobin is often present.
The muscle biopsy often shows an increase in glycogen concentration as determined by periodic acid-Schiff staining. Histological enzyme assays are routinely available for phosphorylase and PFK activity and can confirm a diagnosis in the appropriate clinical context. Electron microscopy usually shows increased glycogen concentration, and in the case of Pompe’s disease, it is membrane bound within lysosomes.
Forearm ischemic, semi-ischemic, and aerobic exercise testing have been described and debated; however, all of these methods demonstrate a failure of lactate to increase with an exaggerated ammonia response (Tarnopolsky et al., 2003). Aerobic exercise testing shows rapid increase in heart rate, rating of perceived exertion, and early fatigue and cramping. If an individual with McArdle’s disease slows down and continues to exercise, there is often a ‘second wind’ that occurs when blood-borne substrates become available and the heart rate falls, and it becomes easier to continue to exercise. The VO2max is often significantly reduced in patients with myopathic GSD.
Magnetic resonance spectroscopy using 31P spectra can demonstrate a lack of acidosis in most cases with distal defects in glycolysis (PFK, phosphoglycerate kinase, phosphoglycerate mutase deficiencies) showing a characteristic increase in phosphomonoesters.
If a specific genetic mutation is known in the family or strongly suspected based on the aforementioned testing, mutation analysis is available for most of the disorders. For example, for a North American Caucasian patient with classic symptoms of muscle cramps with high-intensity exercise and rhabdomyolysis, an initial genetic screen for the most common mutation (R49X) may be prudent.
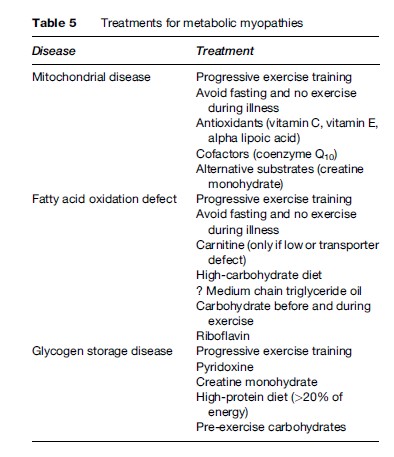
Treatment
Mitochondrial Disorders
The mitochondrial myopathies result in a decrease in aerobic energy transduction through the electron transport chain, an increase in free radical production, and an increased reliance on alternative energy stores. Consequently, therapeutic strategies have traditionally focused on these areas. Strategies that have been used to either bypass specific defects or enhance the flux through the electron transport chain have included; succinate and riboflavin to bypass complex I; coenzyme Q10 as an antioxidant and electron acceptor from complex I and II; antioxidants such as vitamin E, vitamin C, a-lipoic acid and coenzyme Q10. In an attempt to provide an alternative energy supply, some have tried oral creatine monohydrate supplementation with variable success. Studies using dichloroacetate to reduce lactate levels have not been rewarding, and a major side effect of peripheral neuropathy has been seen. Consequently, its use is only advocated during acute severe events with very high lactate concentration. Reviews of nutritional therapies and pharmacological therapies can be found in the following references (Mahoney et al., 2002; DiMauro et al., 2006).
Studies have demonstrated improvement in function in patients following an endurance exercise training program and there is theoretical evidence that higher intensity weight training can activate satellite cells and lessen the mutational burden particularly in sporadic mitochondrial cytopathies (Taivassalo and Haller, 2005). There are a variety of genetic strategies that have been attempted in vitro; however, to date these are not applicable to the clinical scenario (Dimauro et al., 2006).
Fatty Acid Oxidation Defects
The mainstay of therapy is to avoid exercise during the fasting state or during periods of superimposed infection. If an individual has a flu-like illness with vomiting and diarrhea and cannot consume carbohydrates, careful monitoring is required, sometimes with emergency admission and intravenous treatment with glucosecontaining fluids. Most patients can avoid problems with more frequent, higher-carbohydrate feedings, and in some more severe cases cornstarch at night may be required. The consumption of a high-carbohydrate diet including carbohydrate loading and carbohydrates before and during exercise is a mainstay of therapy. Although riboflavin and medium-chain triglyceride oil have been advocated, their efficacy has not yet been demonstrated conclusively in a randomized double-blind trial.
Glycogen Storage Disease
Lifestyle adjustment is probably the best treatment for GSDs, including avoiding brief bursts of high-intensity activity. Interestingly, progressive aerobic conditioning improves functional capacity in such cases, likely by improving fitness and ‘raising the bar’ for the exercise metabolic crisis threshold (Haller et al., 2006). The consumption of sucrose or glucose 15 to 20 min prior to exercise can provide an exogenous source of glucose and improve exercise capacity in patients with glycogenolytic defects (Vissing and Haller, 2003). In contrast, patients with defects in glycolysis do worse with glucose ingestion, because it inhibits lipolysis and they can’t take advantage of the exogenous glucose (due to the block in glycolysis). Pyridoxine (vitamin B6) supplementation has been advocated in the specific entity of McArdle’s disease due to the fact that it is stored in conjunction with the enzyme phosphorylase (which is missing in this entity); however, it has not been evaluated by a rigorous clinical trial. High-protein diets have also been advocated to up-regulate alternative fuel utilization; again, randomized double-blind trials have not been completed. One randomized double-blind trial did demonstrate benefits from the use of creatine monohydrate in low to moderate doses with an impairment of exercise capacity in higher doses (see two papers by Vorgerd and colleagues).
A summary of the treatment strategies for metabolic myopathies can be found in Table 5.
Bibliography:
- DiMauro S, Hirano M, and Schon EA (2006) Approaches to the treatment of mitochondrial diseases. Muscle and Nerve 34(3): 265–283.
- Haller RG, Wyrick P, Taivassalo T, and Vissing J (2006) Aerobic conditioning: An effective therapy in McArdle’s disease. Annals of Neurology 59(6): 922–928.
- Mahoney DJ, Parise G, and Tarnopolsky MA (2002) Nutritional and exercise-based therapies in the treatment of mitochondrial disease. Current Opinion in Clinical Nutrition and Metabolic Care 5(6): 619–629.
- Naviaux RK (2004) Developing a systematic approach to the diagnosis and classification of mitochondrial disease. Mitochondrion 4: 351–361.
- Taivassalo T and Haller RG (2005) Exercise and training in mitochondrial myopathies. Medicine and Science in Sports and Exercise 37(12): 2094–2101.
- Tarnopolsky MA (2004) Exercise testing as a diagnostic entity in mitochondrial myopathies. Mitochondrion 4(5–6): 529–542.
- Tarnopolsky MA (2006) What can metabolic myopathies teach us about exercise physiology? Applied Physiology, Nutrition, and Metabolism 31(1): 21–30.
- Tarnopolsky MA and Raha S (2005) Mitochondrial myopathies: Diagnosis, exercise intolerance, and treatment options. Medicine and Science in Sports and Exercise 37(12): 2086–2093.
- Tarnopolsky M, Stevens L, MacDonald JR, et al. (2003) Diagnostic utility of a modified forearm ischemic exercise test and technical issues relevant to exercise testing. Muscle and Nerve 27(3): 359–366.
- Tein I (1996) Metabolic myopathies. Seminars in Pediatric Neurology 3(2): 59–98.
- Vissing J and Haller RG (2003) The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. New England Journal of Medicine 349(26): 2503–2509.
- Vockley J, Singh RH, and Whiteman DA (2002) Diagnosis and management of defects of mitochondrial beta-oxidation. Current Opinion in Clinical Nutrition and Metabolic Care 5(6): 601–609.
- Vorgerd M, Grehl T, Jager M, et al. (2000) Creatine therapy in myophosphorylase deficiency (McArdle disease): a placebocontrolled crossover trial. Archives of Neurology 57(7): 956–963.
- Vorgerd M, Zange J, Kley R, et al. (2002) Effect of high-dose creatine therapy on symptoms of exercise intolerance in McArdle disease; double-blind, placebo-controlled crossover study. Archives of Neurology 59(1): 97–101.
- Quinlivan R and Beynon RJ (2004) Pharmacological and nutritional treatment for McArdle’s disease (glycogen storage disease type V). Cochrane Database of Systematic Reviews 3: CD003458.
- World Muscle Society (2004) NMD gene tables. Neuromuscular Disorders 14(1). http://www1.elsevier.com/homepage/sah/nmd/ doc/genetables.html (accessed August 2007).
See also:
Free research papers are not written to satisfy your specific instructions. You can use our professional writing services to buy a custom research paper on any topic and get your high quality paper at affordable price.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality



