
This sample Wilson’s Disease Research Paper is published for educational and informational purposes only. If you need help writing your assignment, please use our research paper writing service and buy a paper on any topic at affordable price. Also check our tips on how to write a research paper, see the lists of health research paper topics, and browse research paper examples.
Historical Perspective
Alexander Kinnier Wilson, an American-born neurologist working in Great Britain in 1912, first described progressive lenticular degeneration. He described four cases of a rare, familial, progressive, and invariably fatal disease involving young patients manifested by involuntary movements, spasticity, dysphagia, dysarthria, and psychiatric symptoms (Wilson, 1912). The disorder is characterized by bilateral and symmetrical softening of the lenticular nucleus and by cirrhosis of the liver. Wilson remarked that the disease is likely to be due to ‘‘a toxin associated with the hepatic cirrhosis and generated in connexion therewith.’’
The first description of a patient with Wilson’s disease (WD) was presented in the treatise by Frerichs in 1860 in a child with clinical and pathological features identical to those later described by Wilson (N. Gitlin, 1998). Rumpel first made the relationship between excess copper and the liver in 1913 (Loudianos and Gitlin, 1991). In 1929, Vogt and then Haurowitz and Glazebrook reported on the excess of copper in the brain and liver of patients dying from WD (Stremmel et al., 1991). The excess copper in the cornea, or Kayser Fleischer ring was demonstrated by Policard et al. in 1936 (Kayser, 1902; Stremmel et al., 1991). Finally, Bennetts and Chapman make the relationship between copper metabolism abnormalities and WD after the demonstration of excessive urinary copper excretion.
Sternlieb and Gitlin made the finding of an almost universal low level of ceruloplasmin in the sera of patients with WD in 1952 (Sternlieb, 1980). This then permitted the diagnosis of as yet asymptomatic patients and established an objective method for confirming the clinical impression of the disease.
The genetic basis for WD is linked to one of the transport proteins. The genetic abnormality was linked to the long arm of chromosome 13 by the identification of the association of Wilson’s disease with esterase D deficiency in an Arab inbred family (Bowcock et al., 1987). Subsequent analysis revealed a defect in a P-type ATPase involved in the transport of copper across the trans-Golgi and into transport vesicles. Since then, more than 100 mutations of the ATP7B gene have been identified, which has permitted work on the genotype–phenotype correlation and prenatal screening (Adachi et al., 1997; Riordan and Williams, 2001).
Copper Metabolism And The Pathophysiology Of Wilson’s Disease
Copper is an essential metal for the function of a variety of enzymes. Trace amounts of copper are required for enzymatic reactions involving connective tissue (lysyl oxidase), free radical scavenging (superoxide dismutase), electron transfer (cytochrome c oxidase), pigment production (tyrosinase), and neurotransmission (monamine oxidase) (Stremmel et al., 1991; N. Gitlin, 1998). It is also involved in connective tissue formation (lysyl oxidase) and iron homeostasis.
Foods rich in copper include chocolate, nuts, and shellfish. The average daily intake of copper from the gastrointestinal tract is 1.5–5 mg/day, with the majority of copper absorbed in the stomach and the duodenum. Excess copper intake induces the production of a metallothionein in the enterocyte that captures copper. This allows the safe disposal of excess copper through normal shedding of senescent cells (Sternlieb, 1980; Loudianos and Gitlin, 1991).
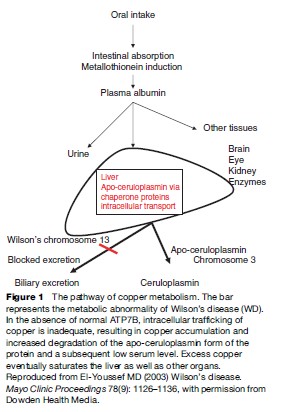
Copper is taken up by albumin and the liver has a high affinity for copper-bound albumin. More than 90% of the copper bound to albumin is present in the liver within a short period after absorption. In the hepatocytes, copper is bound to a thiol-rich cytosolic protein and to specific copper enzymes. Copper is also incorporated into apoceruloplasmin, which has the ability to bind copper atoms. Holoceruloplasmin is a ceruloplasmin protein with a full complement of copper bound to the metal-binding motifs. The incorporation of copper into apo-ceruloplasmin depends on the action of a P-type ATPase that is located in the trans-Golgi and termed ATP7B (Danks, 1991; Gollan and Gollan, 1998). The protein has six metal-binding motifs in its N-terminal domain. This protein directs the incorporation of copper into both apo-ceruloplasmin and the lysosomes. The copper-rich ceruloplasmin is then released in the circulation with about 10% of the copper in the body bound to it. The only physiologic route of copper egress is through biliary secretion. Mutations in this protein product are presumed to result in abnormal accumulation of copper in the liver and an inability of the cell to direct and mobilize the metal. The low ceruloplasmin level is presumed to be caused by rapid intracellular and extracellular degradation of apoceruloplasmin (Riely, 1984b; Brewer and Yuzbasiyan-Gurkan, 1992; Lazaridis and Kamath, 1997; Zucker and Flieder, 1997).
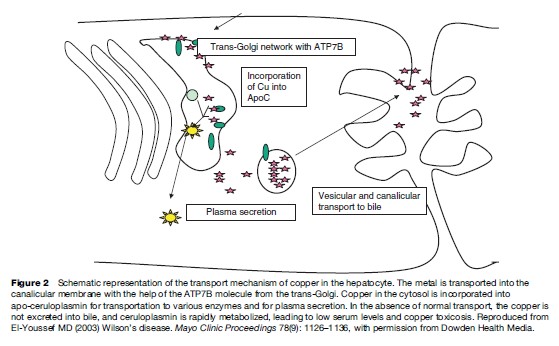
The ATP7B protein is expressed in the trans-Golgi network. It transports copper into the secretory pathway for subsequent incorporation into ceruloplasmin and into a vesicular compartment near the canalicular membrane. The copper in the vesicular compartment is excreted into bile and the ATP7B is redistributed back to the Golgi.
The mechanism of copper incorporation into the ATP7B ATPase depends on the function of cuproproteins, which act as chaperon proteins for trafficking of intracellular copper. The mutations in ATP7B result in abnormal interaction between the ATPase and the chaperon proteins and result in accumulation of copper in the cytoplasm (Brewer and Yuzbasiyan-Gurkan, 1992; Payne et al., 1998).
Ceruloplasmin
Ceruloplasmin is a multi-copper oxidase involved in the oxidation of selected substrates. It is abundant in the serum and contains a substantial proportion of copper in plasma. The synthesis of holoceruloplasmin occurs in the hepatocyte, and the half-life of the protein is estimated to be 5 days. Ceruloplasmin is an acute-phase reactant and its level increases in cases of inflammation, infection, and trauma. About 10% of the synthesized ceruloplasmin is secreted from the liver without copper and is degraded rapidly with a half-life of approximately 5 hours.
Low ceruloplasmin level occurs in WD and in heterozygote patients with aceruloplasminemia. In this later category, there is no associated abnormal copper-related liver pathology. The hepatocytes have, however, a marked increase in iron content ( J.D. Gitlin, 1998). Other cases of low ceruloplasmin occur in newborns, infants in the first 6 months of life, patients suffering from protein-losing enteropathy, and patients with liver cirrhosis of other causes (Sokol et al., 1994).
Clinical Aspects
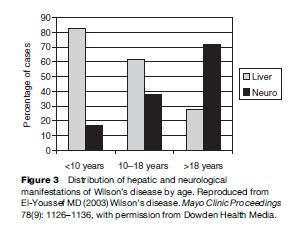
The original description by Wilson emphasized the neurological aspects of the disease. Since then, it has become clear that the neurological manifestations occur in late stages and reflect the accumulation of copper after the liver has been saturated. Neurological manifestations of the disease invariably follow liver involvement, even if it were a silent and unrecognized disease.
It is important to consider the diagnosis in patients who have little if any evidence of the disorder and present with subtle and nonspecific manifestations. A high index of suspicion for WD should be considered in the following circumstances, especially in adolescents and young adults under the age of 40:
- Patients with elevation of liver enzymes found incidentally or in the context of an acute hepatitis episode;
- Patients with dysphagia and dysarthria that is not explained by other neurological disorders;
- Patients with tremors and movement disorders;
- Patients with psychiatric symptoms and liver disease;
- Adolescents with mood disorders and minor elevations of liver tests;
- Patients with Coombs-negative hemolytic anemia;
- Patients with liver cirrhosis;
- Patients with fulminant hepatic failure.
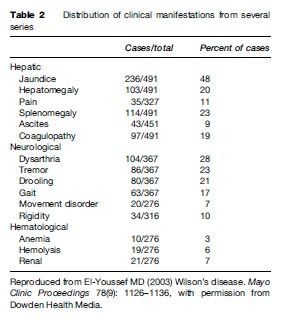
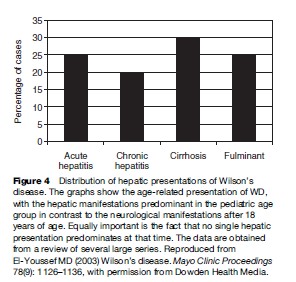
Liver disease is the most frequent initial manifestation of WD. Overall, it accounts for close to 40% of the newly diagnosed cases of WD. Neurological manifestations are the second most likely presenting features. Involvement of more than one organ system at presentation occurs in a quarter of the cases.
Hepatic Aspects
Liver manifestations of WD range from asymptomatic elevation of enzymes to fulminant hepatic failure (Scott et al., 1978; Zitelli et al., 1983; Sternlieb, 1984, 1988; Nazer et al., 1986; Scholsky et al., 1991; Brewer and YuzbasiyanGurkan, 1992; Sokol et al., 1994; Santos Silva et al., 1996; J.D. Gitlin, 1998; Kenngott and Bilzer, 1998; Ferlan-Marolt and Stepec, 1999; Kreymann et al., 1999; Yuce et al., 2000). In fact, WD can mimic a variety of liver conditions, including autoimmune hepatitis, steatosis with or without hepatitis, cirrhosis, and fulminant hepatic failure. A significant proportion of patients present between 3 and 18 years of age and, therefore, WD presentation is a pediatric one. A recent report of a 3-year-old child with acute hepatitis presentation show that WD can present in the preschool years (Wilson et al., 2000). Therefore, any child with liver disease manifestation after age 3 should be screened for WD. Patients are also diagnosed by screening when a family member is identified. Hepatic manifestations of WD occur at a younger age than manifestations of other organ systems. As Table 1 shows, in contrast to neurological manifestations, the age at presentation for hepatic disease occurs in children and young adolescents a general trend of clinical manifestations that may be related to variations of disease severity depending on genetic mutations.

The following are hepatic presenting features of WD:
- Asymptomatic elevation of the liver enzymes found on routine blood testing; this can be minor elevations of one to two times the upper limit of normal (Nazer et al., 1986);
- Bleeding diathesis with subsequent demonstration of end stage liver disease from cirrhosis, including a first esophageal variceal bleed;
- Portal hypertension with hypersplenism presenting as a hematological problem;
- Chronic hepatitis with jaundice and malaise (Scholsky et al., 1991);
- Acute hepatitis characterized by the sudden onset of jaundice, hemolysis, anorexia, and fatigue; the episode lasts 1–2 weeks and is often considered of viral etiology;
- Fulminant presentation with severe coagulopathy and encephalopathy.
Fulminant Hepatic Failure And Hemolysis
Fulminant failure can be the presenting feature of WD (Sternlieb, 1988; Kenngott and Bilzer, 1998; Yuce et al., 2000). Often the diagnosis is not entertained until the presumed viral hepatitis etiology is excluded. This serious complication can occur suddenly. There are some features that permit the differentiation of fulminant failure of WD from other causes: a high level of bilirubin that is out of proportion to the modest elevations of the transaminases, evidence of hemolysis, normal or low alkaline phosphatase, and an AST:ALT ratio greater than 4. Fulminant failure is often associated with renal toxicity from excessive copper egress from dying hepatocytes and is associated with tubular dysfunction characterized by glycosuria, hypophosphatemia, and low uric acid. It is noted that fulminant hepatitis with hemolysis occurs more frequently in females than in males.
Intravascular hemolysis is an important component of fulminant WD and is caused by the massive release of copper from dying hepatocytes. The exact mechanism that leads to massive necrosis of the liver is unknown and is presumed to be caused by the accumulation of large quantities of copper in the lysosomes with subsequent degranulation of proteolytic enzymes and then cell death (Scott et al., 1978; Zitelli et al., 1983.; Riely, 1984a; Sternlieb, 1984, 1988; De Bont et al., 1985; Nazer et al., 1986; Saito, 1987; Scholsky et al., 1991; Brewer and Yuzbasiyan-Gurkan, 1992; Sallie et al., 1992; Sokol et al., 1994; Santos Silva et al., 1996; Hung et al., 1997; J.D. Gitlin, 1998; Kenngott and Bilzer, 1998; Payne et al., 1998; FerlanMarolt and Stepec, 1999; Graff et al., 1999; Kreymann et al., 1999; Pfeil and Lynn 1999; Wilson et al., 2000; Yuce et al., 2000).
Fulminant failure from WD carries near 100% mortality unless liver transplantation is performed. Therefore, it is imperative for the clinician to consider WD in the differential diagnosis of any patient with liver failure and to promptly consider him or her for transplantation (Sternlieb, 1988).
Hemolysis is caused by the disruption of the red blood cell membrane and the depletion of glutathione stores. In animal models, it appears that the level of free serum copper will cause hemolysis when it reaches 75 mg/dl (Sokol et al., 1994; Pfeil and Lynn, 1999).
Chronic Hepatitis
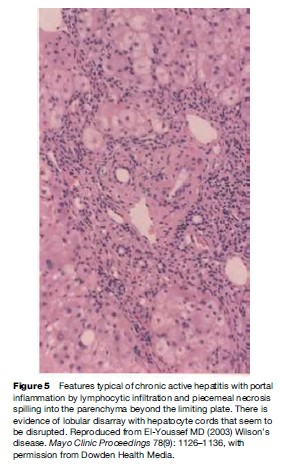
WD can present with features that are very similar to other causes of chronic hepatitis, such as chronic viral hepatitis, autoimmune hepatitis, or drug-related hepatitis (Scott et al., 1978). The characteristic feature that would help distinguish WD from other forms of chronic hepatitis is the mild elevation of the liver enzymes in comparison to the severity of the coagulopathy and to the pathological findings (Saito, 1987). Another important feature is the association with renal tubular dysfunction characterized by low phosphorus and uric acid. Chronic hepatitis with fibrosis and even cirrhosis can present with an acute hepatitis episode that leads to the discovery of chronic liver injury.
Tables 1 and 2 summarize the clinical findings of WD from several series. With variations of reporting, not all findings were uniformly described, hence the differences in totals.
Pathology
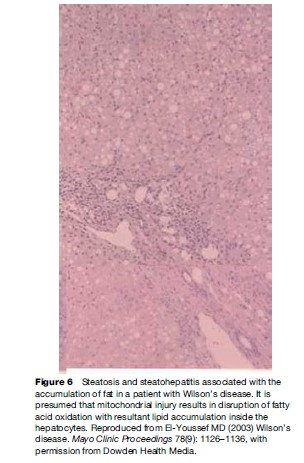
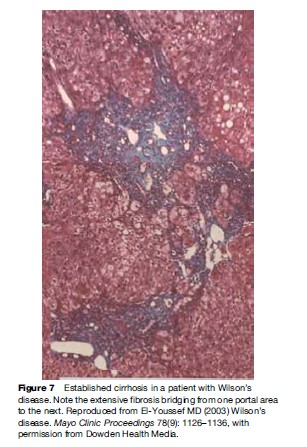
Pathological features of liver involvement in WD can be variable. There is no one feature that is pathognomonic for the diagnosis of WD on liver histology. Even a rhodamine stain for copper is not sufficient to determine the diagnosis of WD on histological grounds alone. Liver histology can be indistinguishable from autoimmune hepatitis, with periportal inflammation, piecemeal necrosis, and lobular disarray (Figure 1). These features can be seen in autoimmune and drug-related hepatitis. Another feature is steatohepatitis, with inflammation and macrovesicular as well as microvesicular steatosis (Figure 2). The accumulation of fat is presumed to be caused by oxidative injury to the mitochondria with subsequent alteration of lipid metabolism. The hepatitis is presumed to result from lipid peroxidation and generation of reactive species and depletion of glutathione. Again, microvesicular steatosis can be seen in hepatitis C and in other metabolic liver diseases in children. In fulminant hepatitis, massive hepatocyte loss is of course the hallmark of liver injury. The differential diagnosis must be made with viral and drug-related hepatitis causes of fulminant failure. Finally, the liver histology may show significant fibrosis and/or cirrhosis by the time the patient comes to medical attention (Figure 3).
Neurologic Aspects
Neurologic manifestations of WD tend to be one of two presentations. The original description by Wilson emphasized the lenticular degeneration aspects of the disease, and these tend to occur at a younger age (Topaloglu et al., 1990; Willeit and Kiechl, 1991; Gow et al., 2000). The pseudosclerosis type of presentation is more common in adulthood. Lenticular degeneration is associated with dystonia and is thought to be less responsive to chelation therapy, whereas the dysarthria and tremors associated with the later onset of pseudosclerosis tend to be more amenable to therapy (Walshe and Yealland, 1992). The frequency of the neurological presentations is outlined in Figures 3 and 4. The frequency of combined dysphagia and dysarthria warrants prompt evaluation for copper toxicosis in any patient that presents with both dysarthria and an abnormal oral phase of the swallowing mechanism (Topaloglu et al., 1990).
Patients may have an exclusive psychiatric presentation. In up to 20% of patients, psychiatric symptoms dominate. They range from depression to phobias, compulsive and antisocial behaviors, to schizophrenic personality disorder (Walshe and Yealland, 1992; Giacchino et al., 1997).
In children, the neurological manifestations of the disease are rare before age 10, with dystonia being the most common feature. Dysarthria, tremors, dysphagia, and psychiatric disturbances do occur in the second decade of life. Insidious dementia manifesting as antisocial behavior, impulsivity, and decreased intellectual performance can be detected with psychometric testing. In 1987, Saito reported on 283 cases of WD, showing that jaundice was the most frequent presenting symptom. In order of decreasing frequency, neurological manifestations were tremors, dysarthria, clumsiness, drooling, and gait disturbance (Saito, 1987).

Computed tomography is more often abnormal in symptomatic than asymptomatic patients. It shows ventricular dilatation and hypodense areas from loss of tissue due to copper toxicity. The affected areas are variable and may show cortical, brainstem, basal ganglia, and posterior fossa hypodensity and atrophy. Positron emission tomography and magnetic resonance imaging may offer more specific signal abnormalities as more patients are evaluated with these modalities.
Eye
Yellow-brown discoloration of Descemet’s membrane in the limbic area of the cornea is termed Kayser-Fleischer ring. Its presence occurs in 98% of patients with neurological disease and in 80% of all cases of WD. The accumulation of copper in this area can be demonstrated best by slit lamp examination but can be visible to the naked eye as well. Kayser–Fleischer rings are not pathognomonic for WD but can be seen in other cases of cholestatic liver disease such as primary biliary cirrhosis, autoimmune hepatitis, and intrahepatic cholestasis associated with prolonged parenteral nutrition. Sunflower cataracts are greenish-gray and are seen by ophthalmoscopic examination of the anterior lens. They usually resolve with chelation therapy (Loudianos and Gitlin, 1991; Stremmel et al., 1991; N. Gitlin, 1998).
Hematological Aspects
Hepatobiliary bilirubinate stones can be the result of hemolysis. Hemolysis can be massive in the context of fulminant failure or as a manifestation of chronic hepatitis (Figures 5 and 6). Hemolysis can be chronic and isolated or in combination with portal hypertension and splenomegaly. The patient may present initially to the hematology service before being recognized as having liver disease (Rosenfield et al., 1978).
Renal Aspects
Renal manifestations of WD involve the glomerulus and the tubules. There is variable azotemia occurring in as many as 20% of patients. Reduction in the glomerular filtration rate has been reported to vary from 10 to 14%. From a review of the literature, it is not clear whether the changes in the glomerular filtration rate is a primary result of copper toxicity or is caused by the renal disease associated with cirrhosis or both (Figure 7). Tubular disease is more clearly related to copper excess as evidenced by the usual improvement with chelation therapy. The spectrum of tubular dysfunction varies from increased uricosuria to Fanconi syndrome with aminoaciduria, tubular acidosis, and various electrolyte abnormalities. These can result in nephrocalcinosis. The loss of glucose in the urine can compound the hypoglycemia of liver failure (Bearn et al., 1957).
Other Aspects
Heart
Cardiomyopathy and congestive heart failure have been described as well as conduction abnormalities and are presumably the result of copper excess. Electrocardiographic changes have shown left ventricular hypertrophy, premature ventricular contractions, atrial fibrillation, and sinoatrial block. Pathological examination has shown ventricular fibrosis and dilated cardiomyopathy (Stremmel et al., 1991).
Endocrine System
Hypoparathyroidism is one complication of WD. The deposition of copper is one possible explanation. The endocrine manifestations of cirrhosis are another cause of metabolic disturbances that are not necessarily directly related to copper toxicity. Amenorrhea and testicular atrophy appear to result from copper toxicity and not necessarily from cirrhosis (Loudianos and Gitlin, 1991; Stremmel et al., 1991; N. Gitlin, 1998).
Muscle
Rhabdomyolysis has been described and may be attributed to the toxicity of copper to mitochondrial function (Loudianos and Gitlin, 1991; N. Gitlin, 1998).
Bones And Joints
Arthritis of the large joints is caused by copper deposition in the synovium. Osteoporosis and osteochondritis dissecans may also occur. Vitamin D-resistant rickets can be the result of renal dysfunction (N. Gitlin, 1998).
Genetics
Wilson’s disease is an autosomal recessive disorder of copper metabolism that occurs in 1:30 000 individuals. It is equally distributed in all ethnic groups and occurs worldwide.
In 1987, genetic evaluation of an Arab family identified several members to be suffering from WD and from a mutation in the red cell enzyme esterase D (Bowcock et al., 1987). This established the location of WD on chromosome 13. Subsequent multi-point linkage analysis identified the abnormal gene to 13q14-q21. Several groups later identified the WD gene independently: it consists of a transcript of 7.5kB expressed primarily in the liver, kidney, and placenta. It is expressed in other tissues at lower levels. The mutations in the WD gene result in a frequency of 1:30 000 persons. The genetic mutation is estimated to occur in 1:90 persons (Adachi et al., 1997; Loudianos et al., 1999; Riordan and Williams, 2001).
The gene codes for a trans-Golgi P-type ATP transport protein. The protein has several metal-binding domains, an ATP-binding domain, a cation channel, and a phosphorylation region, and finally a transduction domain. The protein helps channel copper to the canalicular membrane of the hepatocyte to allow for the biliary excretion of excess copper (Figure 8). The copper is also incorporated into apo-ceruloplasmin for transport to other sites. When apo-ceruloplasmin is not bound to copper it is degraded intracellularly.
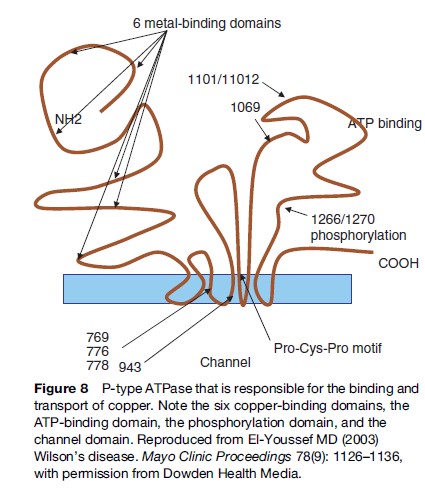
To date, more than 100 mutations have been detected (Adachi et al., 1997; Riordan and Williams, 2001). Most are missense mutations. The most common mutation involves a Histidine ! Glutamine mutation at position 1069. This mutation is responsible for about 40% of cases (Kemppainen et al., 1997; Pyeritz, 1997; Ivanova-Smolenskaya et al., 1999). Whether the variability of the disease affects the phenotypic expression is subject to speculation. Some early reports suggest that some mutations are associated with early onset of disease (Petrukhin et al., 1993; Thomas et al., 1995; Shah et al., 1997). Mutations that result in missense reading seem to cause a slow and progressive hepatic and neurological deterioration, whereas mutations that result in lack of protein product result in early and possibly fulminant hepatic presentation. On the other hand, variability of presentation occurring in the same family suggests other factors that are operative in disease manifestation.
The Histidine → Glutamine mutation was studied in a cell line demonstrating that this mutation results in abnormal folding of the protein and its misdirection to the endoplasmic reticulum with subsequent rapid degradation. Therefore, mutations may result in defective production, defective folding, defective transport function, defective intracellular traffic, and defective chaperon protein interaction (Schaefer et al., 1999; Hamza et al., 2001).
Genetic testing can be used on family members of an index case. The patient’s DNA is used as a reference to recognize the disease-carrying chromosome in other members of the family. Screening of these family members using haplotype analysis with closely linked markers provides precise carrier detection with a 1 to 2% margin of error. The slow and tedious process of genetic testing renders it unsuitable for routine detection of ill patients. The more traditional methods of biochemical testing are more suitable to identify index cases and to screen family members. Genetic testing is available at a few research laboratories (Gaffney et al., 1992; Maier-Dobersberger et al., 1997). Table 3 illustrates the biochemical abnormalities used for the diagnosis of WD (Perman et al., 1979; Walshe, 1988; Martins Da Costa et al., 1992; Cauza et al., 1997).
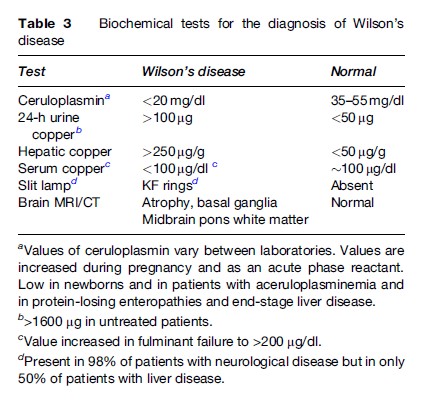
Although the genetic basis of WD is now established, the presence of more than 100 mutations renders genetic testing rather cumbersome, especially in fulminant presentations. Elevated liver copper occurs in cholestatic liver disease such as biliary cirrhosis and autoimmune hepatitis, but other features can help distinguish these conditions from WD.
The measurement of ceruloplasmin using immunoassays includes both holo-ceruloplasmin and apo-ceruloplasmin. Ceruloplasmin being an acute phase reactant may have its value increased during acute illness to levels that would be considered in the normal range. Thus, up to one-third of patients with WD may have a normal ceruloplasmin value. Conditions associated with low ceruloplasmin have been mentioned in the section titled ‘Ceruloplasmin.’ One point is that cirrhosis from other causes can result in a low ceruloplasmin value. In this situation, tissue copper determination may be the only way to determine the diagnosis of WD.
Therapy And Outcome
The rarity of the disease makes it difficult to conduct double-blind placebo-controlled studies of sufficient statistical power to compare one regimen with another. The myriad presentations of WD make it equally difficult to measure outcomes in all major areas of presentation: neurologic, hepatic, and presymptomatic (Walshe, 1996).
D-Penicillamine
Historically, the recognition of the copper-depleting effect of penicillamine led to its adoption as the first choice of chelation in treating copper excess. For more than three decades now, it has been clear that D-penicillamine is capable of reversing the hepatic, neurologic, and psychiatric manifestations of the disease in the majority of patients. It is therefore considered the gold standard of therapy against which other treatments are measured. Asymptomatic patients can be treated effectively and indefinitely, provided they remain compliant. D-Penicillamine is effective in treating patients with severe liver insufficiency who are free of encephalopathy (Durand et al., 2001).
Although D-penicillamine is a chelating agent, reports of fulminant failure occurring in patients who stop the medication abruptly cast doubt on the ability of the drug to de-copper the liver. It is possible that the mechanism of D-penicillamine is multifactorial and may involve induction of intestinal metallothionein and/or act through complex formation in the cell with excess copper (Walshe and Dixon, 1986).
The patient takes 1 to 2 g per day in four divided doses 30 minutes before meals. Rapid mobilization of copper and increased excretion in the urine is expected. Slow and progressive amelioration of liver enzymes follows, and it may take up to 1 year for complete resolution of hepatitis. Likewise, histological improvement occurs in the inflammatory changes and less so with the fibrosis and portal hypertension.
A syndrome of acute neurological deterioration has been observed after initiation of therapy. This may occur in as many as 20% of the patients. In this case, the dose should be reduced to 250 mg/day. Pyridoxine supplementation at a dose of 25 mg should be initiated since D-penicillamine has a weak antipyridoxine effect (Van Caillie-Bertrand et al., 1985; Walshe and Dixon, 1986; Walshe, 1996; Durand et al., 2001).
Trientine
Triethylene tetramine dihydrochloride is used as an alternative therapy for patients who are intolerant to penicillamine (Dubois et al., 1990). Some consider trientine as a first-choice therapy because of its lower incidence of adverse effects. The exact mechanism of action is unknown. The usual dose is 1 to 2 g per day in three divided doses. Sideroblastic anemia is the major side effect of therapy. Most of the few but serious side effects of penicillamine, including nephritis and arthritis, subside with trientine therapy (Dahlman et al., 1995).
Zinc
Zinc’s principal mode of action is presumed to be the induction of metallothionein in the intestine and the liver, thereby sequestering it. Studies have not been able to address the issue of zinc monotherapy as an effective decoppering agent alone since most patients had received other chelating agents. One study suggests continued accumulation of hepatic copper despite zinc therapy. Zinc therapy is efficacious in presymptomatic patients, but more data are needed to address its long-term efficacy. Zinc can be used after chelation has been achieved and as monotherapy in the rare patient who develops intolerance to the both penicillamine and trientine (Brewer et al., 1983; Hoogenraad, 1998; Brewer, 1999).
Thiomolybdates
Tetrahydromolybdate is an investigational drug and has not been approved by the FDA. It is considered particularly suited for the treatment of neurologic manifestations of WD because it is not associated with exacerbation upon initiation of treatment. It interferes with copper absorption and binds to plasma copper. One side effect is bone marrow suppression and another is copper deficiency (Loudianos and Gitlin, 1991; N. Gitlin, 1998).
Liver Transplantation
Liver transplantation (Zitelli et al., 1983; Sternlieb, 1988) is critical for the patient with fulminant hepatic failure. In this setting, the use of living related transplantation results in copper metabolism that is similar to patients who are heterozygote genetic carriers, since a parent is often the donor (Komatsu et al., 2002). It is also recommended for cirrhotic patients with end-stage liver disease. It is generally not recommended for the patient suffering from neurological disease without significant liver deterioration. Hepatic manifestations are reversed by liver transplantation. Neurological manifestations of the disease may or may not reverse with transplantation, but the number of cases is only anecdotal.
Recently, hepatocyte transplantation was performed in Long-Evans Cinnamon rats, which model WD perfectly, with encouraging results showing not only normalization of histology, an indicator of the potential of transplanted cells to repopulate and regenerate the liver, but also biliary excretion of copper was restored to normal (Irani et al., 2001).
Conclusion
WD has evolved from a uniformly fatal disease to an eminently treatable condition. Our understanding of the disease has moved from the clinical description to the biochemical and histological aspects, and finally to genetic basis of copper metabolism. As always, each advance poses new questions and challenges in diagnosis and management. Wilson’s disease should be considered in the differential diagnosis of any patient with abnormal liver function tests, in patients with fulminant hepatic failure, and in patients with an associated constellation of neurologic, hematologic, and hepatic disease. It is also important to remember that the disease is one of children and adolescents, and therefore the possibility of a serious metabolic cause for a child with liver disease should consider WD in its differential diagnosis. Finally, recognition of WD in any patient should prompt the evaluation of the family and genetic counseling. It is imperative to initiate and maintain treatment of the asymptomatic patient as soon as the diagnosis is made. It is no easy task to convince a patient in his or her prime to take relatively unpleasant medication for life when the symptoms are not even present.
Bibliography:
- Adachi Y, Okuyama Y, Miya H, and Kamisako T (1997) Presence of ATP-dependent copper transport in the hepatocyte canalicular membrane of the Long-Evans cinnamon rat, an animal model of Wilson-disease. Journal of Hepatology 26(1): 216–217.
- Bearn AG, Yu TK, and Gutman AB (1957) Renal function in Wilson’s disease. Journal of Clinical Investigation 36: 1107.
- Bellary S, Hassanein T, and Van Thiel DH (1995) Liver transplantation for Wilson’s disease. Journal of Hepatology 23(4): 373–381.
- Bowcock AM, Farrer LA, Cavalli-Sforza LL, et al. (1987) Mapping the Wilson disease locus to a cluster of linked polymorphic markers on chromosome 13. American Journal of Human Genetics 41(1): 27–35.
- Brewer GJ (1999) Treatment of Wilson’s disease with zinc. Journal of Laboratory and Clinical Medicine 134(3): 322–324.
- Brewer GJ and Yuzbasiyan-Gurkan V (1992) Wilson’s disease. Medicine (Baltimore) 71(3): 139–164.
- Brewer GJ, Hill GM, Prasad AA, Cossack ZT, and Rabbani P (1983) Oral zinc therapy for Wilson’s disease. Annals of Internal Medicine 99(3): 314–319.
- Cauza E, Maier-Dobersberger T, Polli C, Kaserer K, Kramer L, and Ferenci P (1997) Screening for Wilson’s disease in patients with liver diseases by serum ceruloplasmin. Journal of Hepatology 27(2): 358–362.
- Chowrimootoo GF, Andoh J, and Seymour CA (1997) Western blot analysis in patients with hypocaeruloplasminaemia. Quarterly Journal of Medicine 90(3): 197–202.
- Dahlman T, Hartvig P, Lofholm M, Nordlinder H, Loof L, and Westermark K (1995) Long-term treatment of Wilson’s disease with triethylene tetramine dihydrochloride (trientine). Quarterly Journal of Medicine 88(9): 609–616.
- Danks DM (1991) Copper and liver disease. European Journal of Pediatrics 150(3): 142–148.
- De Bont B, Moulin D, Stein F, Van Hoof F, and Lauwerys R (1985) Peritoneal dialysis with D-penicillamine in Wilson disease. Journal of Pediatrics 107(4): 545–547.
- Dubois RS, Rodgerson DO, and Hambidge KM (1990) Treatment of Wilson’s disease with triethylene tetramine hydrochloride ( Trientine). Journal of Pediatric Gastroenterology and Nutrition 10(1): 77–81.
- Durand F, Bernuau J, Giostra E, et al. (2001) Wilson’s disease with severe hepatic insufficiency: Beneficial effects of early administration of D-penicillamine. Gut 48(6): 849–852.
- El-Youssef MD (2003) Wilson’s disease. Mayo Clinic Proceedings 78(9): 1126–1136.
- Ferlan-Marolt V and Stepec S (1999) Fulminant Wilsonian hepatitis unmasked by disease progression: Report of a case and review of the literature. Digestive Diseases and Sciences 44(5): 1054–1508.
- Gaffney D, Walker JL, O’Donnell JG, et al. (1992) DNA-based presymptomatic diagnosis of Wilson disease. Journal of Inherited Metabolic Disease 15(2): 161–170.
- Giacchino R, Marazzi MG, Barabino A, et al. (1997) Syndromic variability of Wilson’s disease in children: Clinical study of 44 cases. Italian Journal of Gastroenterology and Hepatology 29(2): 155–161.
- Gitlin JD (1998) Aceruloplasminemia. Pediatric Research 44(3): 271–276.
- Gitlin N (1998) Wilson’s disease: The scourge of copper. Journal of Hepatology 28(4): 734–739.
- Gollan JL and Gollan TJ (1998) Wilson’s disease in 1998: Genetic, diagnostic and therapeutic aspects. Journal of Hepatology 28 (supplement 1): 28–36.
- Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, and Sewell RB (2000) Diagnosis of Wilson’s disease: An experience over three decades. Gut 46(3): 415–419.
- Graff C, Clayton DA, and Larsson NG (1999) Mitochondrial medicine: Recent advances. Journal of Internal Medicine 246(1): 11–23.
- Hamza I, Faisst A, Prochaska J, et al. (2001) The metallochaperone Atox 1 plays a critical role in perinatal copper homeostasis. Proceedings of the National Academy of Sciences of the United States of America 5: 6848–6852.
- Hoogenraad TU (1998) Zinc treatment of Wilson’s disease. Journal of Laboratory and Clinical Medicine 132(4): 240–241.
- Houwen RH, Juyn J, Hoogenraad TU, Ploos van Amstel JK, and Berger R (1995) H714Q inmutation in Wilson disease is associated with late, neurological presentation. Journal of Medical Genetics 32: 480–482.
- Hung IH, Suzuki M, Yamagachi Y, et al. (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. Journal of Biological Chemistry 272: 21461–21466.
- Irani AN, Malhi H, Slehria S, et al. (2001) Correction of liver disease following transplantation of normal rat hepatocytes into Long-Evans cinnamon rats modeling Wilson’s disease. Molecular Therapy 3(3): 302–309.
- Ivanova-Smolenskaya IA, Ovchinnikov IV, Karabanov AV, et al. (1999) The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease. Journal of Medical Genetics 36(2): 174.
- Kayser B (1902) Ueber einen Fall von angeborener grunlicher Verfarbung der Cornea. Klinische Monatsblatter fur Augenheilkd 40: 22.
- Kemppainen R, Palatsi R, Kallioinen M, and Oikarinen A (1997) A homozygous nonsense mutation and a combination of two mutations of the Wilson disease gene in patients with different lysyl oxidase activities in cultured fibroblasts. Journal of Investigative Dermatology 108(1): 35–39.
- Kenngott S and Bilzer M (1998) Inverse correlation of serum bilirubin and alkaline phosphatase in fulminant Wilson’s disease. Journal of Hepatology 29(4): 683.
- Komatsu H, Fujisawa T, Inui A, et al. (2002) Hepatic copper concentration in children undergoing living related liver transplantation due to Wilsonian fulminant hepatic failure. Clinical Transplantation 16(3): 227–232.
- Kreymann B, Seige M, Schweigart U, Kopp KF, and Classen M (1999) Albumin dialysis: Effective removal of copper in a patient with fulminant Wilson disease and successful bridging to liver transplantation: A new possibility for the elimination of protein-bound toxins. Journal of Hepatology 31(6): 1080–1085.
- Lazaridis KN and Kamath PS (1997) 28-year-old man with renal insufficiency and jaundice. Mayo Clinic Proceedings 72(11): 1057–1060.
- Loudianos G and Gitlin JD (1991) Wilson’s disease. Seminars in Liver Disease 20(3): 353–364.
- Loudianos G, Dessi V, Lovicu M, et al. (1999) Mutation analysis in patients of Mediterranean descent with Wilson’s disease: Identification of 19 novel mutations. Journal of Medical Genetics 36: 833–836.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. (1997) Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Annals of Internal Medicine 127(1): 21–26.
- Martins Da Costa C, Baldwin D, Portmann B, Lolin Y, Mowat AP, and Mieli-Vergani G (1992) Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology 15(4): 609–615.
- Nazer H, Ede RJ, Mowat AP, and Williams R (1986) Wilson’s disease: Clinical presentation and use of prognostic index. Gut 27(11): 1377–1381.
- Payne AS, Kelly EJ, and Gitlin JD (1998) Functional expression of the
- Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069G mutation. Proceedings of the National Academy of Sciences of the United States of America 95: 10854–10859.
- Perman JA, Werlin SL, Grand RJ, and Watkins JB (1979) Laboratory measures of copper metabolism in the differentiation of chronic active hepatitis and Wilson disease in children. Journal of Pediatrics 94(4): 564–568.
- Petrukhin K, Lutsenko S, Chernou I, Ross B, Kaplan J, and Gilliam T (1993) Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nature Genetics 5: 338–343.
- Pfeil SA and Lynn DJ (1999) Wilson’s disease: Copper unfettered. Journal of Clinical Gastroenterology 29(1): 22–31.
- Pyeritz RE (1997) Genetic heterogeneity in Wilson disease: Lessons from rare alleles. Annals of Internal Medicine 127(1): 70–72.
- Riely CA (1984a) Acute hepatic failure in children. Yale Journal of Biological Medicine 57(2): 161–184.
- Riely CA (1984b) Wilson’s disease. Pediatrics in Review 5(7): 217–222.
- Riordan SM and Williams R (2001) The Wilson’s disease gene and phenotypic diversity. Journal of Hepatology 34(1): 165–171.
- Rosenfield N, Grand RJ, Watkins JB, Ballantine TV, and Levey RH (1978) Cholelithiasis and Wilson disease. Journal of Pediatrics 92(2): 210–213.
- Saito T (1987) Presenting symptoms and natural history of Wilson disease. European Journal of Pediatrics 146(3): 261–265.
- Sallie R, Katsiyiannakis L, Baldwin D, et al. (1992) Failure of simple biochemical indexes to reliably differentiate fulminant Wilson’s disease from other causes of fulminant liver failure. Hepatology 16(5): 1206–1211.
- Santos Silva EE, Sarles J, Buts JP, and Sokal EM (1996) Successful medical treatment of severely decompensated Wilson disease. Journal of Pediatrics 128(2): 285–287.
- Schaefer M, Hopkins R, Failla M, et al. (1999) Hepatocyte-specific localization and copper-dependent trafficking of the Wilson’s disease protein in the liver. American Journal of Physiology 276: G639–G646.
- Scholsky ML, Scheinberg IH, and Sternlieb I (1991) Prognosis of Wilsonian chronic active hepatitis. Gastroenterology 100: 762–767.
- Scott J, Gollan JL, Samourian S, and Sherlock S (1978) Wilson’s disease, presenting as chronic active hepatitis. Gastroenterology 74(4): 645–651.
- Shah A, Chernov I, Zang H, et al. (1997) Identification and analysis of mutations in the Wilson disease gene (ATP7B): Population frequencies, genotype-phenotype correlation, and functional analyses. American Journal of Human Genetics 61: 17–28.
- Sokol RJ, Twedt D, McKim JM Jr., et al. (1994) Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. 1. Gastroenterology 107(6): 1788–1798.
- Sternlieb I (1980) Copper and the liver. Gastroenterology 78(6): 1615–1628.
- Sternlieb I (1984) Wilson’s disease: Indications for liver transplants. Hepatology 4(supplement 1): 15S–17S.
- Sternlieb I (1988) Wilson’s disease: Transplantation when all else has failed. Hepatology 8(4): 975–976.
- Stremmel W, Meyerrose K, Niederau C, Hefter H, Kreuzpaintner G, and Strohmeyer G (1991) Wilson Disease: Clinical presentation, treatment, and survival. Annals of Internal Medicine 115: 720–726.
- Thomas G, Forbes J, Roberts E, Walshe J, and Cox D (1995) The Wilson disease gene: Spectrum of mutations and their consequences. Nature Genetics 9: 210–217.
- Topaloglu H, Gucuyener K, Orkun C, and Renda Y (1990) Tremor of tongue and dysarthria as the sole manifestation of Wilson’s disease. Clinical Neurology and Neurosurgery 92(3): 295–296.
- Van Caillie-Bertrand M, Degenhart HJ, Luijendijk I, Bouquet J, and Sinaasappel M (1985) Wilson’s disease: Assessment of D-penicillamine treatment. Archives of Diseases in Childhood 60(7): 652–655.
- Walshe JM (1988) Diagnosis and treatment of presymptomatic Wilson’s disease. Lancet 20: 435–437.
- Walshe JM (1996) Treatment of Wilson’s disease: The historical background. Quarterly Journal of Medicine 89(7): 553–555.
- Walshe JM and Dixon AK (1986) Dangers of non-compliance in Wilson’s disease. Lancet 1(8485): 845–847.
- Walshe JM and Yealland M (1992) Wilson’s disease: The problem of delayed diagnosis. Journal of Neurolology, Neurosurgery and Psychiatry 55(8): 692–696.
- Willeit J and Kiechl SG (1991) Wilson’s disease with neurological impairment but no Kayser-Fleischer rings. Lancet 337(8754): 1426.
- Wilson DC, Phillips MJ, Cox DW, and Roberts EA (2000) Severe hepatic Wilson’s disease in preschool-aged children. Journal of Pediatrics 137(5): 719–722.
- Wilson SAK (1912) Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver. Brain 34: 295.
- Wright T (1991) Role of copper in Wilson’s diseases. Presented to the Contemporary Gastroenterology, July and August 1991.
- Yuce A, Kocak N, Gurakan F, and Ozen H (2000) Wilson’s disease presented with fulminating hepatic failure in children. Digestive Diseases and Sciences 45(4): 704.
- Zitelli BJ, Malatack JJ, Gartner JC, et al. (1983) Orthotopic liver transplantation in children with hepatic-based metabolic disease. Trans Proceedings, No. 1, March 1983.
- Zucker SD and Flieder A (1997) Case records of the Massachusetts General Hospital. Case 1 – 1997 – A 23-year-old man with fulminant hepatorenal failure of uncertain cause. New England Journal of Medicine 336(2): 118–125.
See also:
Free research papers are not written to satisfy your specific instructions. You can use our professional writing services to buy a custom research paper on any topic and get your high quality paper at affordable price.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality



