
This sample Muscular Dystrophies Research Paper is published for educational and informational purposes only. If you need help writing your assignment, please use our research paper writing service and buy a paper on any topic at affordable price. Also check our tips on how to write a research paper, see the lists of health research paper topics, and browse research paper examples.
Introduction
Muscular dystrophy (MD), as described by Walton and Nattrass in 1954, is a heterogeneous group of inherited primary diseases of the muscle, clinically characterized by progressive muscle weakness and wasting. Histologically, it is unified by the presence of necrotic and regenerating processes, often associated with an increased amount of connective and adipose tissues (Emery, 2001). Following this definition, the discussion will be focused on dystrophinopathies, Emery-Dreifuss muscular dystrophies, congenital muscular dystrophies, limb-girdle muscular dystrophies, and fascioscapulohumeral muscular dystrophy. The heterogeneity of these different disorders included is often delineated by a combination of clinical, genetic, molecular, and pathological aspects, which will be thoroughly discussed in this research paper.
Dystrophinopathies
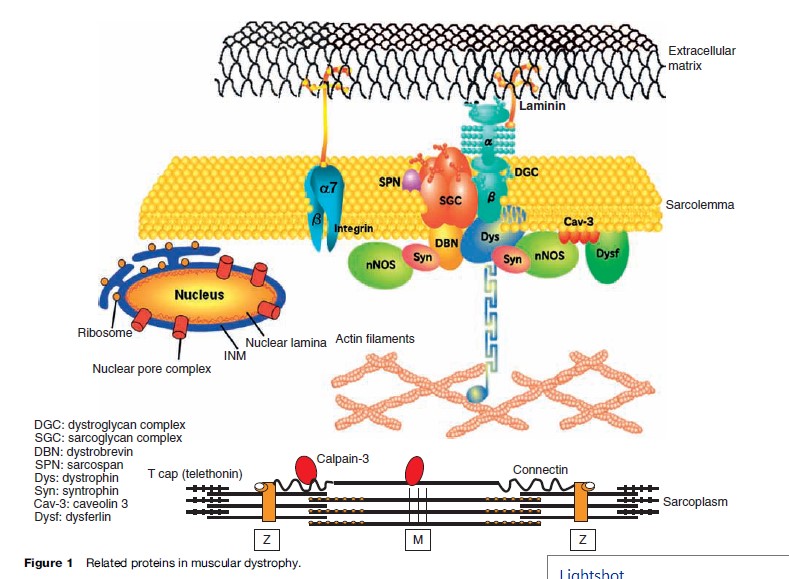
Dystrophinopathies are X-linked progressive hereditary degenerative diseases of skeletal muscles caused by an absence or deficiency of dystrophin, a sarcolemmal protein. Dystrophin and the associated proteins form a complex integral framework connecting the intracellular actin cytoskeleton to the extracellular matrix, stabilizing the sarcolemma from mechanical stresses during muscle contraction (Figure 1). The responsible gene is located on the short arm of the X chromosome at locus Xp21. It is an extremely large gene, comprising more than 2.5 million base pairs and 79 exons. Out-of-frame mutation of dystrophin can result in Duchenne muscular dystrophy (DMD), while in-frame mutation causes its milder allelic form, Becker muscular dystrophy (BMD). In both DMD and BMD, the most common mechanism of mutation is large-scale deletion.
Duchenne Muscular Dystrophy
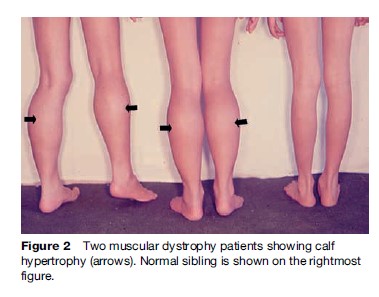
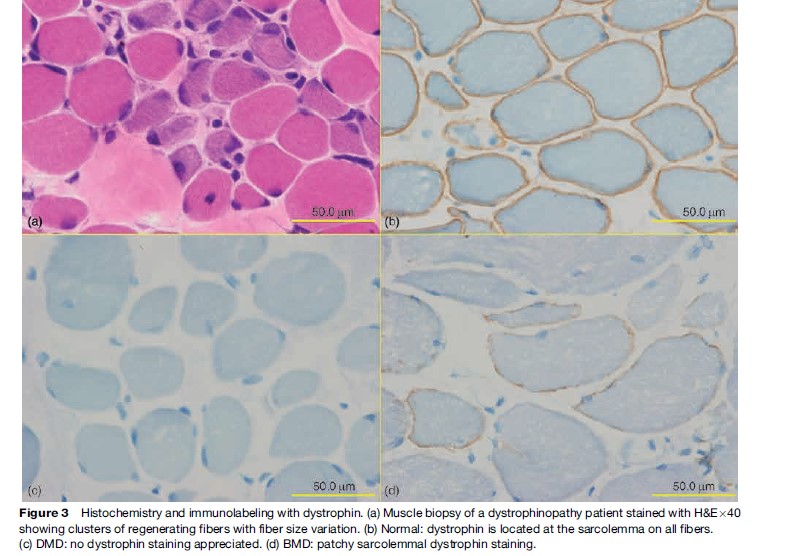
Named after a French physician in 1861, DMD is one of the most common muscular dystrophies occurring in approximately 1 in 5000 male births. Symptoms are usually recognized by 2 years of age when an apparent delayed motor development is noted. In over 50% of cases, walking is delayed until 18 months and affected boys never learn to run normally. Muscle involvement is often bilateral and symmetrical. As knee and hip extensor weakness progresses, the child employs the Gowers’ maneuver, whereby he climbs up his thighs with both hands to extend his hips and trunk in assuming a standing position. In most cases, calf muscle hypertrophy (Figure 2) is observed. Cardiac and central nervous system involvement is clearly evident, with cardiomyopathy and low IQ associated in some cases. Creatine kinase (CK) is usually elevated, with levels greater than ten times the upper normal limits during the early stages; it never reaches a normal level, even during the terminal stage. Pathologically, fiber size variation associated with clusters of necrotic and regenerating processes (Figure 3a) is often observed, with complete absence of dystrophin on immunostaining (Figure 3c). This disease is slowly progressive and most patients are wheelchair-bound by 15 years of age. Previously, death occurred at around 20 years of age due to respiratory failure; however, improvement in respiratory care has prolonged life expectancy. Most patients now live to around 30 years of age and die from cardiac complications. Female DMD has also been reported due to X chromosome translocation involving the dystrophin gene locus. In other instances, Turner syndrome (this syndrome encompasses several conditions, most commonly monosomes X. In females, normally there are XX sex chromosomes, but in this syndrome, there is only one X chromosome which is fully functional. As a result, the karyotype is labeled |45, X|| instead of 46, XX|) is associated with DMD due to complete or partial absence of an X chromosome or nonrandom X chromosome inactivation.
Becker Muscular Dystrophy
Becker muscular dystrophy, named after a well-known German geneticist, occurs in approximately one out of 30 000 male births. Although the same dystrophin gene is mutated, the clinical features of BMD vary greatly from DMD-like phenotype to no subjective weakness. The onset of disease is much later. Muscle cramp and calf hypertrophy are frequently noted. Cardiac abnormalities may also be seen, but mental retardation is rare. Creatine kinase is elevated. Muscle biopsy findings include an active necrotic and regenerative process with endomysial fibrosis. In milder cases, type 2 fiber can be hypertrophic, while type 1 fibers are atrophic. Patchy dystrophin immunostaining is diagnostic (Figure 3d). The presence of some measure of functioning dystrophin in BMD patients allows a more benign course and most affected individuals lose ambulation only after 20–30 years of illness.
Female carriers of both DMD/BMD can also be symptomatic with elevated serum creatine kinase and muscle weakness with mosaic pattern of dystrophin immunostaining. Some carriers develop cardiomyopathy later in life.
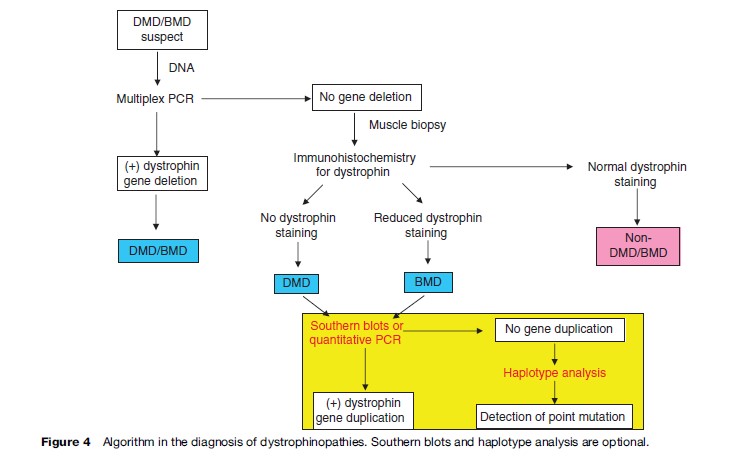
For initial diagnosis of dystrophinopathies, multiplex polymerase chain reaction (PCR) should be performed. This method can detect deletion in 60% in all DMD and 85% in all BMD patients, while Southern blots and quantitative PCR can be used to detect duplication. The algorithm in diagnosis of dystrophinopathies is shown in Figure 4. Prenatal diagnosis could be performed by DNA studies on chorionic villus sampling at 8–10 weeks gestation and cultured amniotic fluid cells at 12–16 weeks gestation.
Emery-Dreifuss Muscular Dystrophy
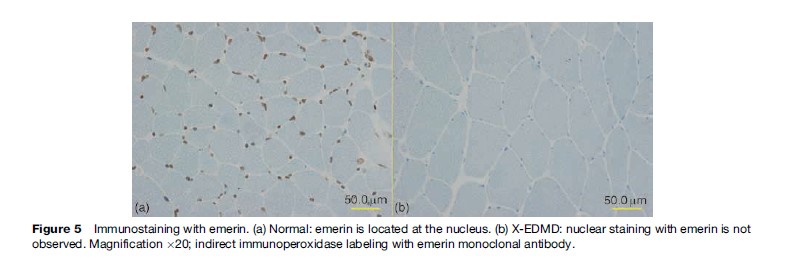
Emery-Dreifuss muscular dystrophy (EDMD) was coined by Rowland in 1979 after the two physicians who detailed, described, and differentiated the disease from DMD/BMD. It is characterized by a triad of early joint contractures, progressive humeroperoneal muscle wasting, and cardiomyopathy with conduction block. There are three modes of inheritance: X-linked recessive (X-EDMD), autosomal dominant (AD-EDMD), and autosomal recessive (AREDMD). X-EDMD is often caused by a nonsense mutation in the EMD gene, which is located on the X chromosome at locus Xq28, resulting in the absence of an inner nuclear protein emerin (Figure 5). The clinical onset of disease is usually in the first or second decade of life with joint contractures involving the elbows, Achilles tendon, and posterior neck appearing before any significant muscle weakness. The nature of the early onset contracture is still not understood at present. Muscle weakness initially involves the humeroperoneal areas and later involving the scapular and pelvic muscles. In most cases of X-EDMD, cardiac abnormalities appear later than muscle involvement, generally after 20–30 years of age. In contrast, both AD-EDMD and AREDMD are included in a group of diseases collectively known as laminopathies, which stem from missense mutation of the Lamin A/C (LMNA) gene located on chromosome 1q21.2-q21.3, resulting in a nonfunctional nuclear lamina protein. As reported by Bonne et al. in 2000, the clinical presentation of the autosomal form, including the age of onset and severity of disease, is much more heterogeneous than X-EDMD. Joint contractures occur after muscle weakness. Cardiac conduction defects in EDMD can manifest as sinus bradycardia, first-degree atrioventricular block, Wenckebach phenomenon, third-degree atrioventricular block, and bundle-branch block. Atrial and ventricular arrhythmias are frequent. In AD-EDMD, the risk of ventricular tachyarrhythmia and dilated cardiomyopathy manifested by left ventricular dilation and dysfunction is higher than in XL-EDMD. Serum CK is moderately elevated. Muscle biopsy often reveals nonspecific features such as fiber size variation with necrotic and regenerating process.
Congenital Muscular Dystrophy
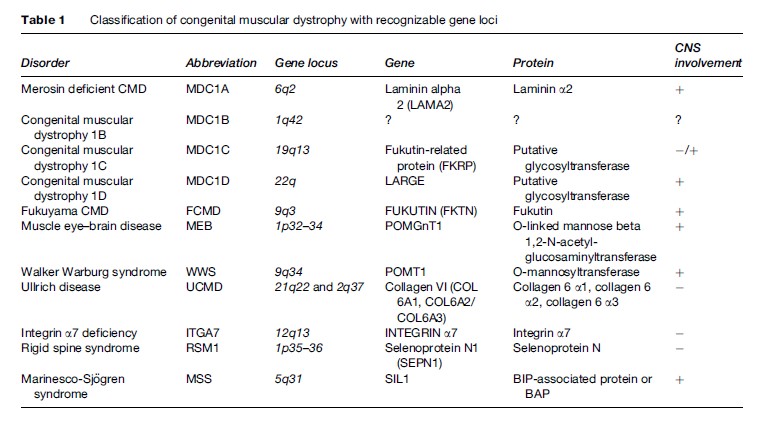
First described by Batten in 1903, congenital muscular dystrophy (CMD) is a group of clinically heterogeneous autosomal recessive inherited muscle diseases characterized by hypotonia at birth, generalized muscle weakness, frequently multiple contractures, and necrotic and regenerating process on muscle biopsy. The clinical spectrum ranges from a very severe form, often resulting in early infant death, to relatively mild conditions, where the patient survives into adulthood. The recent advances of molecular genetics have led to a more comprehensive classification of CMD based on the clinical presentation, genetic abnormality, and the primary biochemical protein defects (Table 1). Presently, nine genes responsible for the different type of CMD have been identified. Importantly, the frequency of each subtype varies widely among different ethnic groups.
Laminin 2 (Merosin) Deficiency
Laminin a2 deficiency comprises 40–50% of all CMD in European countries. However, in Japan, it accounts for only a few percent. Laminin a2, encoded by the LAMA gene, is a glycoprotein in the basement membrane, which binds to a number of macromolecules including agrin, nidogen, and collagen IV in the extracellular matrix and serves as ligand to two transmembrane proteins, alpha dystroglycan and integrin (Figure 1). Laminin a2 is expressed in muscle, cerebral blood vessels, developing white matter tracts, and Schwann cells. Affected children present with hypotonia, weakness, and respiratory and feeding problems at birth or in the first few months of life. Contractures can occur but severe arthrogryposis is rare. Sitting without support is the maximal motor ability attained. Calf hypertrophy and facial muscle weakness can be observed in the neonatal period. Brain magnetic resonance imaging (MRI) invariably shows white matter changes in patients after 6 months, despite normal mentation. However, structural brain anomalies are also observed in some cases, causing mental retardation and epilepsies (Muntoni and Voit, 2004). Demyelination of the motor nerve is also observed in some patients with reduced motor nerve conduction velocity. Serum CK is invariably elevated, usually more than ten times the normal values in the early stages. Failure to thrive occurs in 80% of the cases, from weakness of the muscles involved in swallowing often resulting in aspiration pneumonia. Coupled with frequent infection, respiratory muscle weakness eventually leads to respiratory failure, necessitating ventilatory support. Mild to moderate cardiac hypokinesia is seen in a small portion of MCD1A patients. Immunostaining techniques can readily demonstrate the absent or partial reduction of merosin. However, in the latter cases, further test is warranted for careful interpretation.
Fukuyama Congenital Muscular Dystrophy
Currently, five genes (FKTN, POMGnT1, POMT1, FKRP, and LARGE ) are identified encoding proteins and glycosyltransferases involved in the glycosylation of a-dystroglycan. Collectively, disorders resulting from mutation in these genes are called a-dystroglycanopathies. The common pathologic mechanism governing these diseases is aberrant glycosylation of a-dystroglycan, which might explain the existing phenotypic overlap between the different forms. Immunolabeling reveals complete absence or reduced a-dystroglycan. Among them, Fukuyama congenital muscular dystrophy (FCMD) is caused by mutation in FKTN gene. This disease, originally described by Fukuyama and his collaborators in 1960, is the most frequent CMD in Japan, accounting for 60% of cases. Virtually all FCMD patients are Japanese. This is because only the Japanese share the common 3 kb retrotransposal insertion in the 30 UTR of the FKTN gene. Seventy-five percent of FCMD cases in Japan are attributable to homozygous 3 kb retrotransposal insertion into the 30 UTR of the fukutin gene and 25% have compound heterozygous mutations of the 3 kb insertion at the 30 UTR and a mutation in the coding sequence. The classical features include hypotonia, generalized weakness, facies, mental retardation, and ocular anomalies. Motor improvement is often observed in a brief period between ages 2 and 8 years. Typically, the patient sits independently but never walks. This is followed by progressive weakness with subsequent respiratory failure. Brain changes are often due to abnormal neuronal migration, resulting in profound mental retardation and epilepsy. Dilated cardiomyopathy typically develops in the second decade of life. Life expectancy averages approximately 20 years but with the advent of more sophisticated respiratory equipment, survival into the third decade is increasingly possible. Recently, a patient with a homozygous 1bp insertion was reported from Turkey. This patient had severe brain and eye abnormalities and died 15 days after birth.
Muscle Eye Brain Disease
Muscle eye brain disease (MEB) due to mutation in the POMGnT1 gene was first described in Finland in 1977, but presently, worldwide distribution including Japan and Korea is known. The clinical spectrum of brain and muscle involvement is similar to that described in FCMD patients but typically with more severe eye dysfunction manifested as poor visual alertness during the neonatal period. Despite the loss of glycosylated a-dystroglycan and laminin a-2 binding capacity, the expression of core a-dystroglycan is preserved in MEB.
Walker-Warburg Syndrome
Walker-Warburg syndrome, first described in 1942, is the most severe form of the a-dystroglycanopathy associated with life expectancy of less than 3 years. Although mutation in FKTN and FKRP account for a small fraction of WWS, mutation in POMT1 has also been recently identified in a well-characterized cohort of WWS patients, suggesting genetic heterogeneity. The characteristic feature of marked weakness is compounded by the severe brain and eye anomalies.
Congenital Muscular Dystrophy 1C
Fukutin-related protein (FKRP) is ubiquitously expressed throughout the human body with the highest levels in the skeletal and cardiac muscles. Recently, FKRP gene mutation has been considered to be associated with the widest phenotypic spectrum of muscular dystrophy ranging from in utero onset and early fetal lethality in WWS or MEB to late adult onset, slowly progressive limb-girdle muscular dystrophy (LGMD2I). Mutation in this gene, which is associated with reduced immunolabeling of a-dystroglycan, is likewise responsible for congenital muscular dystrophy 1C (MDC1C). The hallmark is severe muscle weakness and early onset of respiratory insufficiency. Serum CK levels are always elevated (20–75 times). Intelligence and brain MRI are normal. Heart involvement is less prominent, in contrast to LGMD2I. A considerable number of MDC1C/ LGMD2I patients have been identified in European and Middle Eastern countries, but only a few patients have been found in Japan.
Congenital Muscular Dystrophy 1D
Loss of function mutation in the LARGE gene, as depicted in the myodystrophy (Largemyd) mouse, displays a severe, progressive muscular dystrophy and mild cardiomyopathy, in addition to retinal and peripheral and central nervous system involvement. Recently, recessive mutation in the human homolog LARGE gene has been identified to be responsible for congenital muscular dystrophy 1D (MDC1D), which is congenital muscular dystrophy associated with profound mental retardation brought about by white matter changes and subtle structural abnormalities on brain MRI. Pathologically, there is reduced immunolabeling of a-dystroglycan and reduced molecular weight of a-dystroglycan on immunoblot with preserved laminin binding ability.
Ullrich Congenital Muscular Dystrophy
Collagen VI, a ubiquitously expressed extracellular matrix protein, forms microfibrils in close association with the basal lamina around the muscle fibers. This protein interacts with several other matrix constituents. It is composed of three chains, a1, a2, and a3, encoded by the genes COL6A1 and COL6A2 on chromosome 21q22.3 and COL6A3 on chromosome 2q37. Recessive mutation in any of the three genes causes Ullrich congenital muscular dystrophy (UCMD). Originally reported by Ullrich in 1930 as congenital atonic-sclerotic muscular dystrophy, the typical phenotype is represented by a combination of early-onset severe muscle weakness with proximal joint contractures and distal joint hyperextensibility. Normal intelligence is also an integral feature of the disease. Characteristic round facies, lid lag, and prominent ears are also often observed. Progressive functional deterioration is mostly due to increased contractures, which subsequently involves even the distal joint in later stages. Respiratory insufficiency invariably develops in the first or second decade. Serum CK is normal or mildly elevated. Pathological features in the muscle biopsy range from mildly myopathic with occasional necrotic and regenerating fibers to overtly dystrophic with profound endomysial and perimysial fibrosis. Collagen VI immunolabeling reportedly shows three different patterns of abnormalities: Complete deficiency, partial deficiency, and sarcolemmaspecific collagen VI deficiency (SSCD). Complete deficiency is associated with recessive mutation, while SSCD is associated with dominant mutations. An increasing number of patients has recently been recognized. Most likely, this is one of the major CMDs, accounting for 10–20% of CMD cases.
Rigid Spine Syndrome
The clinical phenotype of rigid spine syndrome was initially described by Victor Dubowitz. Following linkage study, Moghadaszadeh et al. identified selenoprotein N gene (SEPN1) as the causative gene for this disease. It encodes an endoplasmic reticulum glycoprotein with the highest expression in the fetal tissues, suggesting its functional role in the developing muscular system. Severely affected patients present as floppy infants with weak suckling, poor neck flexion, and they never achieve ambulation. However, in more mildly affected children, symptoms may present in infancy or childhood with delayed motor development or frequent falls. In such cases, ambulation is usually maintained into adulthood. The hallmark of this syndrome is spinal rigidity and scoliosis due to contractures of the spine extensor muscles, which may develop between the ages of 3 and 12 years. Serum CK is normal or mildly elevated and muscle imaging reveals selective involvement of medial thigh muscles with relative sparing of rectus femoris and gracilis. Vital capacity decreases over time due to the stiffness of the rib cage with compounding diaphragmatic weakness. Pathologically, fiber size variation with mild necrotic and regenerating process, type 1 fiber predominance, and atrophy may be observed. Many specimens would show unevenness of intermyofibrillar network in oxidative staining, sometimes with overt corelike features.
Integrin 7 Deficiency
Integrins are a major laminin a2 receptor in the muscle fibers. The accurate expression and localization of integrin a7b1 is laminin a-2-dependent. Primary deficiency of integrin a7 is rare, and so far causative mutation in ITGA7 has only been identified in three patients. These patients manifested as mild congenital myopathy with delayed motor milestones. Muscle biopsies revealed only mild variation in fiber size. Direct diagnosis through immunolabeling of integrin a7 seems to be hindered by the developmental regulation and interindividual variation observed in the first 2 years of life wherein expression of the protein employing the available antibodies is frequently low.
Marinesco Sjogren Syndrome
Marinesco-Sjoren syndrome is a rare disorder that is inherited as an autosomal recessive genetic condition. The major features of this disorder are cerebellar atrophy causing ataxias, congenital cataracts, and psychomotor retardation. Other cardinal features include myopathies, short stature, and hypergonadotrophic hypogonadism. Muscle pathology is characterized by fiber size variation, necrotic fibers, rimmed vacuoles with marked connective tissue, and fat replacement. The underlying causative gene SIL1 encodes for a co-chaperone for HSPA5 (also called BiP), which is a chaperone for the heat-shock protein 70 (HSP70) (Antonen et al., 2006). Recently, researchers have suggested that disturbed SIL1–HSPA5 interaction and protein folding is most likely the primary pathogenesis in Marinesco Sjo¨ gren syndrome.
Limb-Girdle Muscular Dystrophies
The limb-girdle muscular dystrophies (LGMDs) are a group of genotypically and phenotypically heterogeneous muscular dystrophies (Bushby, 1999). They are characterized by weakness of the proximal muscles in the upper and lower extremities. Involvement is usually first evident in either the pelvic or, less frequently, the shoulder girdle, often with asymmetry of wasting when the upper limbs are first involved. Pathologically, findings include variation in fiber size, dystrophic process, internal nucleation, and endomysial fibrosis.

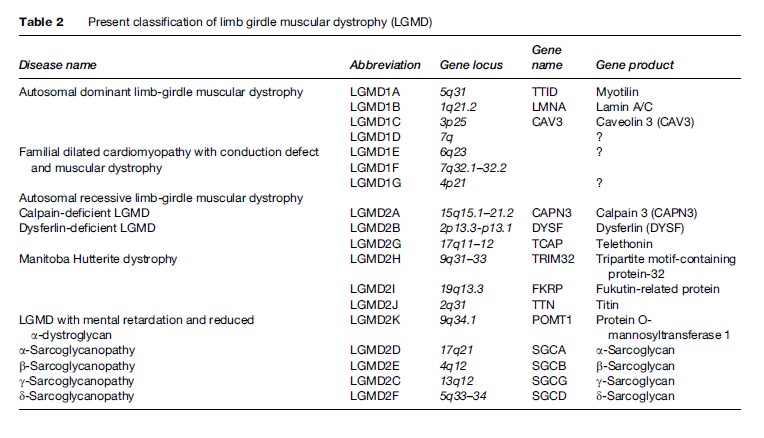
The identification of the DAP complex and the characterization of the complex’s role in the pathogenesis of many LGMDs has provided a new basis for the distinction and classification of these muscular dystrophies (Table 2). Presently, there are 16 different loci identified, seven autosomal dominant causing type 1 LGMD and 14 autosomal recessive causing type 2 LGMD. Further genetic heterogeneity exists for both autosomal dominant and autosomal recessive LGMD, as established by further linkage studies.
LGMD2 cases are more common, having a cumulative prevalence of 1:15 000 with a number of geographical differences. Moreover, these are more severe, with some resembling DMD phenotype. Four of the recessive forms have been associated with defects in genes coding for the sarcoglycan complex, which along with dystrophin helps anchor muscles to the extracellular matrix. More devastating mutations in these same genes can cause severe childhood autosomal muscular dystrophy (SCARMD). In contrast, only a relatively small proportion of LGMD, around 10%, is autosomal dominant in inheritance.
Calpainopathy
Calpainopathy (LGMD2A) involves mutations in the CAPN3 gene, which encodes the muscle-specific protein calpain-3. It is characterized mainly by a symmetric, very selective atrophic involvement of limb-girdle and trunk muscles, with the gluteus maximus and thigh adductors being most affected. Clinically, calf hypertrophy is rarely observed, but Achilles tendon contractures are common. Scapular winging is usually present from the early stages, though it may be asymptomatic. Onset of symptoms occurs between the ages of 8 and 15 years, but adult onset is not uncommon. Progression of calpainopathy is variable. Most patients may have normal mobility in childhood with a very slowly progressive course of disease. Confinement to a wheelchair occurs at the earliest typically 11–28 years after onset. Anterior distal leg and distal arm muscles are relatively spared. While the first location of detectable muscle weakness was in the pelvifemoral muscles, muscle CT scanning in some patients showed early involvement of the gastrocnemius muscle, which was usually asymptomatic, though early inability to walk on tiptoe is an important clinical clue. Respiratory, but not cardiac, complications have been reported.
The majority of patients with CAPN3 mutations had a variable degree of calpain-3 protein deficiency determined by immunoblot analysis. The probability of having LGMD2A is high when patients have a complete calpain-3 deficiency and progressively decreases with increasing amounts of protein detected. Patients with severe, early-onset disease usually have no calpain-3 protein, but absent or markedly reduced protein levels are also detected in patients with adult onset. However, almost all patients with normal calpain-3 levels have late or adult onset of the disorder. Given the heterogeneity of the mutations seen in this very large gene, muscle biopsy will probably still prove a more straightforward starting point for diagnosis than mutation detection, though this will remain necessary for carrier detection and prenatal diagnosis where this is required.
Dysferlinopathy (LGMD2B)
Dysferlinopathy (LGMD2B) is secondary to mutations in the dysferlin gene. In this disease, CK levels are often 20–150 times above the normal range at presentation. Disease onset usually is in the late teens, and progression of illness is slow. Commonly, patients have inability to stand on tiptoes because of the predominant distal lower limb involvement. Dysferlin localizes to the muscle fiber membrane and is expressed from very early in human development. This gene is involved in two forms of muscular dystrophy: LGMD2B and the predominantly distal muscular dystrophy, Miyoshi myopathy. The underlying means by which mutations in this gene are responsible for these different phenotypes is not known. Interestingly, both forms (LGMD2B and Miyoshi) can co-exist in the same family.
Sarcoglycanopathies
There are five known sarcoglycans (a, b, g, d, and possibly e) (Figure 1) at the muscle fiber membrane that forms part of the DAPC, and genetic defects affecting four (a-, b-, g-, and d-sarcoglycan) of these proteins cause specific type 2 LGMDs, also known as sarcoglycanopathies (LGMD2C, 2D, 2E, and 2F). The most characteristic clinical features of this group include calf hypertrophy, involvement of scapular and deltoid muscles at onset of disease, and cardiomyopathy. Symptoms mostly occur during childhood, although the onset can occur in adulthood. Progression of the disease ranges from very mild to very severe.
a-Sarcoglycanopathy (LGMD2D) patients commonly present with difficulty running and climbing stairs; like in dystrophinopathy, however, they can present with muscle cramps or exercise intolerance. Toe-walking is an early feature in fewer than half of the patients. Other patients have delayed walking, implying an earlier onset, but adult-onset cases have also been reported. Calf hypertrophy is seen in almost all patients at some stage. Facial, ocular, and velopharyngeal muscles are spared. Progression of the disease is usually faster with earlier presentation, but some intrafamilial variability has been observed.
g-Sarcoglycanopathy (LGMD2C) patients have similar presentation as those having LGMD2D. They have proximal lower limb weakness, frequent calf hypertrophy, variability in severity, which may be intrafamilial, predominantly with childhood onset, no cardiac involvement, and normal intelligence (Noguchi et al., 1995).
b-Sarcoglycanopathy (LGMD2E) patients with this myopathy have a broad range of clinical severity, even within the same family. The mean age at onset usually is at 7–8 years. Patients with an adult onset usually have a milder phenotype; ambulation is still possible in some patients in the sixth decade. More severe forms have earlier onset during childhood and faster progression.
Patients with d-sarcoglycanopathy (LGMD2F) tend to show a very severe clinical course. Age at onset ranges from 4 to 10 years, with confinement to a wheelchair between 9 and 16 years and death between 9 and 19 years.
Limb-Girdle Muscular Dystrophy 2G
Limb-girdle muscular dystrophy 2G (LGMD2G) has been shown to be secondary to mutations in the gene encoding telethonin. Patients typically present early in the second decade with difficulty climbing stairs and running, although foot-drop is also an early feature. Proximal and distal lower-limb weakness is therefore present from the onset, while in the upper limbs the proximal musculature is more severely affected. Confinement to a wheelchair occurs approximately 18 years after onset. Creatine kinase may be mildly or moderately elevated. Muscle biopsies show a considerable number of rimmed vacuoles.
Limb-Girdle Muscular Dystrophy 2H
Limb-girdle muscular dystrophy 2H (LGMD2H), linked to chromosome 9q31–33, has been shown to be caused by mutations in the gene encoding a tripartite motif-containing protein-32 (TRIM32) among Hutterites. The initial report was from Jerusalem et al. in 1973, where two brothers from an inbred Hutterite colony with nonprogressive weakness from infancy were diagnosed as having sarcotubular myopathy. Affected individuals presented with proximal lower limb weakness between 8 and 27 years of age, with elevated serum creatine kinase concentration (2–30 times normal). Facial and proximal upper limb muscles, predominantly the trapezius and deltoid were affected as disease progressed. Mild distal limb muscle (brachioradialis and anterior peroneal muscle) involvement was also seen. Most patients have remained ambulant late into adult life.
Limb-Girdle Muscular Dystrophy 2I
Limb-girdle muscular dystrophy 2I (LGMD2I) is caused by mutation in the gene encoding fukutin-related protein (FKRP). Age at onset ranges from 6 months to 40 years. The disorder can be phenotypically variable. Patients can present with hypotonia, waddling gait, or difficulty in climbing stairs, weakness in the hip and shoulder girdle muscles, calf hypertrophy, tight Achilles tendon, and elevated serum creatine kinase. Young boys can resemble DMD or BMD phenotype. Some patients show signs of cardiomyopathy on electrocardiogram. In the consanguineous Tunisian family originally reported by Driss et al. (2000), however, the patients had symmetric proximal muscle weakness and wasting in all four limbs but no heart involvement.
Limb-Girdle Muscular Dystrophy 2J
Limb-girdle muscular dystrophy 2J (LGMD2J) is caused by homozygous mutation in the titin gene (TTN). Heterozygous mutation in the titin gene causes tardive tibial muscular dystrophy (TMD). Mutation in the titin gene also causes dilated cardiomyopathy type 1G (CMD1G). As reported by Udd et al. in a consanguineous Finnish pedigree, 8 out of 20 members had a severe limb-girdle muscular dystrophy inherited in an autosomal recessive pattern, with an onset in the first to third decades involving all proximal muscles, although some patients developed distal muscle involvement. Severe disability with loss of ambulation occurs on the third to sixth decades. There was no facial muscle involvement or cardiomyopathy. The findings were compatible with the hypothesis that the severe LGMD phenotype was the homozygous manifestation of a dominant gene that in the heterozygous state caused the milder distal myopathy. In patients with homozygous mutations, complete loss of calpain-3 is seen, which may be a secondary downstream effect of deficiency of the TMD gene protein and results in phenotypic overlap with LGMD2A.
Limb-Girdle Muscular Dystrophy 2K
Limb-girdle muscular dystrophy 2K (LGMD2K), caused by mutation in the gene encoding protein O-mannosyltransferase-1 (POMT1), is an early-onset myopathy, which is allelic to Walker-Warburg syndrome, and is mostly found in Turkish families. All patients acquire early motor milestones, excluding congenital muscular dystrophy. Age at onset ranges from 1 to 6 years, with difficulty in walking and climbing stairs. Other features include slow progression, proximal muscle weakness, mild muscle hypertrophy, increased serum creatine kinase, microcephaly, and mental retardation.
Limb-Girdle Muscular Dystrophy 1A
Limb-girdle muscular dystrophy 1A (LGMD1A), secondary to mutations in the myotilin-encoding gene, is an autosomal dominant disorder characterized by adult onset of proximal muscle weakness, beginning in the hip girdle and later progressing to the shoulder girdle region. Distal muscle weakness may occur later. Progression of the disease is very slow, and very few patients progress to wheelchair confinement.
Limb-Girdle Muscular Dystrophy 1B
Limb-girdle muscular dystrophy 1B (LGMD1B) is caused by LMNA mutations. In affected individuals, symmetric weakness starts in the proximal lower-limb muscles before the age of 20 years. In the third or fourth decade, upper-limb muscles gradually become affected as well. Early contractures of the spine are absent, and contractures of elbows and Achilles tendons are either minimal or late, distinguishing this disorder from Emery-Dreifuss muscular dystrophy. Like EDMD, however, cardiac involvement is also prominent, mostly in the form of atrioventricular conduction blocks. A minority of patients also have dilated cardiomyopathy (van der Kooi et al., 1997). While cardiac problems very rarely precede muscle symptoms, in some patients their muscle symptoms are recognized only in retrospect.
Limb-Girdle Muscular Dystrophy 1C
Limb-girdle muscular dystrophy 1C (LGMD1C) is an autosomal dominant muscular dystrophy, associated with a severe deficiency of caveolin-3 in muscle fibers (up to 95% reduction). The clinical features are usually mild, comprising calf hypertrophy and muscle pain on exertion. Age of onset is around 5 years and disease progression is variable, as patients can remain ambulant up to adulthood. Episodes of exertional muscle cramps are noted in some patients. Serum creatine kinase levels are elevated fourto 25-fold.
The remaining LGMDs are only defined by a chromosomal localization since the genes associated with these dystrophies have not yet been cloned. LGMD1D has been mapped to chromosome 7q. Onset is between the second and sixth decades, with hip girdle preceding shoulder girdle involvement. Creatine kinase is elevated and muscle biopsy shows fiber splitting and fibrosis. LGMD1E, mapped to 6q23, characteristically is associated with cardiomyopathy, conduction defects, with or without muscle weakness. It is also referred to as dilated cardiomyopathy 1F (CMD1F). It has been identified in a large family of
French-Canadian descent. LGMD1F, linked to chromosome 7q32.1–32.2, has been described in one Spanish family. Proximal muscle weakness is seen during the onset of the disease, involving distal muscles in the later stage. Muscle biopsy revealed myopathic changes and sometimes rimmed vacuoles. LGMD1G, mapped to chromosome 4p21, was described in a Brazilian Caucasian family. Clinical presentation is rather homogenous: patients present with cramps early in the course, gradually developing proximal myopathy. Contractures produce limited flexion of fingers and toes.
Despite the recent advances in the molecular characterization of LGMDs, a clear method of classification is not yet available. In the future, when all the causative genes and their respective protein products are identified, a new method of classification may be feasible.
Facioscapulohumeral Muscular Dystrophy
Facioscapulohumeral (FSH) muscular dystrophy is an autosomal dominant form that affects muscles of the face (facio), scapula (scapulo), and upper arms (humeral). Although extraocular muscles are not affected, weakness in muscles around the eye (i.e., orbicularis oculi) may be evident when patients sleep with their eyes slightly open, a symptom that may manifest itself before other symptoms develop. There is also difficulty in whistling or blowing. Distal extremity muscles also can be affected, and the weakness is often asymmetric. There is a wide range of clinical severity, with symptoms that can include cardiac, cognitive, visual, and auditory impairments. Symptoms may develop in early childhood and are usually noticeable in teenage years. Life expectancy is normal, but some affected individuals become severely disabled. Nearly all cases are associated with a distal 4q35 deletion.
Clinical diagnosis of FSHD is based initially on the pattern of muscle involvement, but genetic tests, which can detect FSHD with a 98% success rate, are now preferred. Because there are no known genes in this region, a novel position effect has been postulated to explain the disease phenotype. Although the specific gene responsible for FSHD has not been identified, FSHD is associated with a deletion of 3.3-kb repeats (called D4Z4) mapped to the telomeric end of the long arm of chromosome 4. This region, termed 4q35, normally has 11–150 D4Z4 repeats, but in patients with FSHD, the number of repeats is fewer than 11. Although it was thought until recently that the severity of clinical presentation increased as the number of D4Z4 repeats under 11 decreased, this has recently been challenged. The telomeric D4Z4 repeats have most recently been thought to affect anchoring of chromosome 4 to the inner nuclear membrane. If true, FSHD would resemble other MDs, such as Emery-Dreifuss muscular dystrophy, that are nuclear in origin.
Bibliography:
- Antonen A, Mahjneh I, Ha¨ ma¨ la¨ inen R, et al. (2006) The gene disrupted in Marinesco Sjo¨ gren syndrome encodes SIL1, an HSPA5 cochaperone. Nature Genetics 37: 1309–1311.
- Bonne G, Mercuri E, Muchir A, et al. (2000) Clinical and molecular genetic spectrum of autosomal dominant muscular dystrophy due to mutations of the lamin A/C gene. Annals of Neurology 48: 170–180.
- Bushby KM (1999) The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Human Molecular Genetics 8(10): 1875–1882.
- Driss A, Amouri C, Hamida CB, et al. (2000) A new locus for autosomal recessive limb girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscular Disorders 10: 240–246.
- Emery A (2001) The Muscular Dystrophies. New York: Oxford University Press.
- Muntoni F and Voit T (2004) The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscular Disorders 14: 635–649.
- Noguchi S, McNally EM, Ben Othmane K, et al. (1995) Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science 270: 819–821.
- van der Kooi AJ, van Meegen M, Ledderhof TM, et al. (1997) Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. American Journal of Human Genetics 60: 891–895.
- Bonnenmann CG and Bushby KM (2004) The limb girdle muscular dystrophies. In: Engel AG and Franzini-Armstrong C (eds.) 3rd edn. 1077–1121. Myology, 3rd edn., pp. 1077–1121. New York: McGraw-Hill.
- Hayashi YK (2005) X-linked form of Emery-Dreifuss Muscular dystrophy. Acta Myologica 24: 98–103.
- Nishino I and Ozawa E (2002) Muscular dystrophies. Current Opinion in Neurology 15: 539–544.
- Voit T and Tome F (2004) The congenital muscular dystrophies. In: Engel AG and Franzini-Armstrong C (eds.) 3rd edn. 1203–1238. Myology, 3rd edn., pp. 1203–1238. New York: McGraw-Hill.
See also:
Free research papers are not written to satisfy your specific instructions. You can use our professional writing services to buy a custom research paper on any topic and get your high quality paper at affordable price.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality



