
This sample Transmissible Spongiform Encephalopathies Research Paper is published for educational and informational purposes only. If you need help writing your assignment, please use our research paper writing service and buy a paper on any topic at affordable price. Also check our tips on how to write a research paper, see the lists of health research paper topics, and browse research paper examples.
Introduction
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are a group of rapidly progressive, uniformly fatal brain degenerative diseases that occur in humans and animals. Neuropathologic characteristics common to most TSEs include neuronal loss, spongiform lesions, astrogliosis, and absence of inflammatory reaction. The presence of protease-resistant prion protein can be demonstrated in the brain and occasionally in other tissues of humans and animals affected by TSEs. Acquired forms of TSEs are characterized by long incubation periods, usually measured in years. Purification of a hydrophobic protein presumed to be the causative agent of TSEs was first reported by Prusiner and colleagues (1982). In 1997, Dr. Stanley Prusiner was awarded the Nobel Prize for his contribution in the discovery and characterization of the TSE agent.
TSEs in humans include kuru, Creutzfeldt-Jakob disease (CJD), variant CJD, Gerstmann-Stra¨ussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI) (Table 1). Kuru was identified in the 1950s among the Fore tribe of Papua New Guinea and may have affected over 3000 patients, primarily women and children, before virtually disappearing after cessation of the ritualistic cannibalism, which facilitated its transmission (Will et al., 2004). TSEs that occur in animals include scrapie, bovine spongiform encephalopathy (BSE, also called mad cow disease), chronic wasting disease (CWD), transmissible mink encephalopathy (TME), feline spongiform encephalopathy, and ungulate spongiform encephalopathy (Table 1). Feline and ungulate spongiform encephalopathies represent transmission of BSE to domestic cats and zoo animals, respectively. Scrapie in sheep and goats, first reported in the 1700s in Europe, occurs endemically worldwide with the probable exception of Australia and New Zealand. TME has occurred in outbreaks among ranched mink in Canada, Finland, Germany, Russia, and the United States. The last known TME outbreak occurred in 1985 in Wisconsin. TSEs were relatively obscure diseases, but they were catapulted into wider recognition in the 1990s after the emergence of BSE and its presumed transmission to humans attracted much media and public attention.
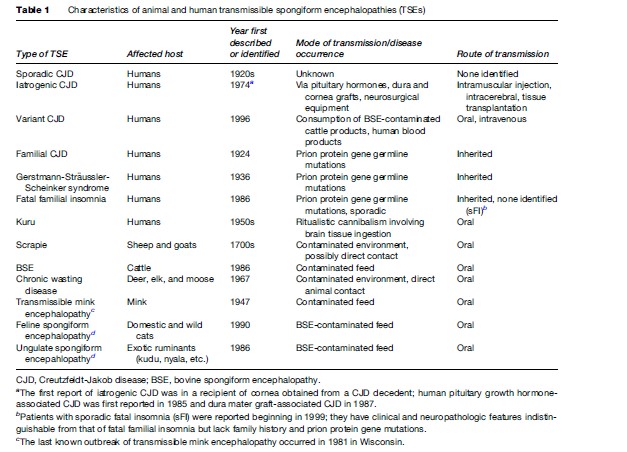
Causative Agent Of TSEs
Several agents had been proposed as possible causative agents of TSEs. Before the 1980s, the most widely implicated agents were ‘slow viruses.’ However, no viral particles or disease-specific nucleic acids were identified in association with scrapie transmission in laboratory animals. Replication of the scrapie agent in the absence of nucleic acids and its possible proteinaceous nature were postulated as early as the 1960s by Alper and colleagues, Pattison and Jones, and Griffith. The protein-only hypothesis began to be widely accepted after Prusiner and colleagues purified a hydrophobic protein and demonstrated that the presence of this protein was required for scrapie transmission in laboratory animals. In 1982, Prusiner coined the colloquial term prion to describe this protein by joining the first few letters from the descriptive phrase ‘proteinaceous infectious’ particle. Since then, additional evidence has accumulated indicating that prions may actually be acting alone in causing TSEs. However, some critics of the protein-only hypothesis suggest that nucleic acids undetected by current methods may play a crucial role in the pathogenesis of TSEs.
Prions appear to be composed largely or entirely of a protein designated as PrPSc and are abnormal conformers of a host-encoded cellular protein designated as PrPC. PrPC is a structural component of cell membranes primarily in the brain but also in other tissues of humans and other mammals. Although its function is unknown, it may be involved in supporting neuronal synaptic activity and copper binding and may also interact with other cell-surface proteins to render neuroprotective functions. In humans, PrPC is encoded by the prion protein gene located on the short arm of chromosome 20.
The fundamental event in the occurrence of TSEs appears to be conversion of the cellular PrPC into the abnormally folded, pathogenic PrPSc. This conversion appears to be dictated by the presence of PrPSc and occurs by a poorly defined posttranslational autocatalytic process, requiring the aid of cofactors such as proteins or nucleic acids. The initial PrPSc molecule may originate from exogenous sources or within the brain from somatic or germline prion protein gene mutations. Knockout mice devoid of PrPC do not develop a TSE even after inoculation with infectious prions, indicating that expression of the prion protein gene is a prerequisite for generation and propagation of PrPSc. Conversion to PrPSc seems to confer more beta sheet structure and resistance to proteolytic enzymes, conventional disinfectants, and standard sterilization methods. The cellular PrPC is sensitive to denaturation by these chemical and physical methods, and its structure is primarily composed of alpha helices.
Creutzfeldt-Jakob Disease
CJD, the most common form of TSE in humans, bears the name of two German neurologists who reported patients with rapidly progressive degenerative brain diseases in the early 1920s. Neuropathologic review of Creutzfeldt’s initial patient was inconclusive despite having the typical CJD clinical manifestations. In contrast, two of the four patients reported by Jakob had the typical neuropathologic features of CJD (Belay, 1999).
In most patients, CJD is characterized by the onset of dementia, ataxia, or behavioral abnormalities between the ages of 55 and 75 years. As the disease progresses, neurologic dysfunction deteriorates and patients develop speech abnormalities, movement disorders, such as myoclonus and akinetic mutism (Will et al., 2004). Typically, the disease progresses rapidly, and over 50% of the patients die within 6 months and about 80% within 1 year of illness onset. Duration of illness longer than 2 years is extremely rare. A characteristic electroencephalogram (EEG) finding of triphasic, periodic sharp waves can be demonstrated in about 75% of patients. In the appropriate clinical context, elevated cerebrospinal fluid (CSF) levels of 14-3-3 protein may also aid in diagnosing CJD. More recently, abnormal brain magnetic resonance imaging (MRI) findings in the basal ganglia and cortical regions have been reported to be suggestive of a CJD diagnosis. However, these MRI findings and CSF 14-3-3 protein elevation have been demonstrated in other brain diseases mimicking CJD. A definitive CJD diagnosis requires histopathologic or immunodiagnostic testing of brain tissues obtained at autopsy or biopsy. Histopathologic examination of brain tissue from CJD patients shows the hallmark triad of spongiform lesions, neuronal loss, and astrogliosis. Immunodiagnostic testing, such as immunohistochemistry and Western blot analysis, demonstrates the presence of PrPSc, confirming a diagnosis of CJD.
CJD occurs in three different forms: a sporadic form with no known environmental source of infection, an iatrogenic form accidentally transmitted via medical interventions, and a familial form associated with prion protein gene mutations. Decades of research have not identified a specific source of infection for sporadic CJD, which accounts for about 85% of patients. Spontaneous generation of PrPSc was hypothesized as a cause for sporadic CJD, possibly resulting from random somatic mutations or errors during prion protein gene expression. Iatrogenic CJD has resulted from receipt of contaminated cadaveric human pituitary hormones and dura mater and corneal grafts and from exposure to contaminated neurosurgical equipment. In addition, beginning in the mid-1990s, a newly recognized variant form of CJD linked with BSE transmission to humans was reported.
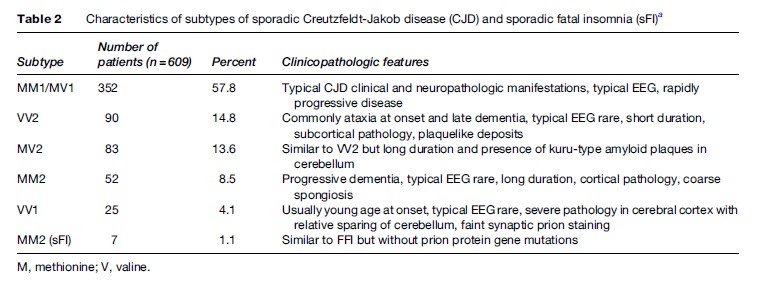
Sporadic CJD can be further subdivided into five different subtypes based on the Western blot characteristics of protease-resistant PrPSc fragment and the polymorphism at codon 129 of the host prion protein gene (Table 2) (Parchi et al., 1999). The different subtypes correlate with specific clinical and neuropathologic phenotypes. The most common subtype is associated with prion type 1 and the presence of methionine at codon 129 of the prion protein gene.
Bovine Spongiform Encephalopathy
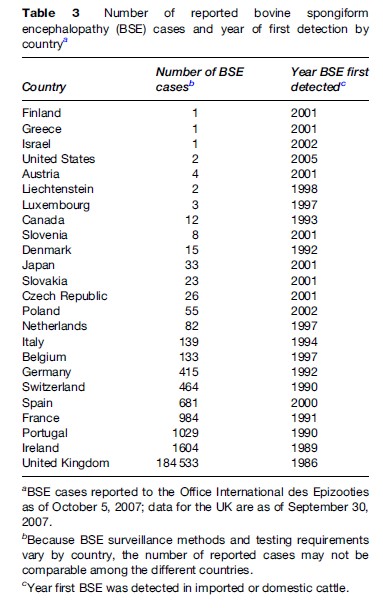
BSE was first recognized in 1986 in the United Kingdom where it caused a large outbreak among cattle (Table 3). Earlier BSE cases were retrospectively identified from 1985, and a modeling study suggested that cases might have occurred since the early 1980s. The number of UK BSE cases increased almost exponentially, peaked during 1992–1993, and markedly declined thereafter. The clinical signs of BSE include unsteady gait with falling, behavioral abnormalities, and abnormal responses to touch and sound. Because of the fearful and aggressive behavior exhibited by some of the earlier BSE-infected cattle, the public media used the term mad cow disease to describe the strange disease affecting many cattle in the UK. This term continues to be used, sometimes erroneously, to describe human TSEs such as CJD.
The original source of the BSE outbreak is unknown. The leading hypotheses include spontaneous occurrence of BSE and scrapie transmission to cattle from feed containing rendered, infected sheep carcasses. The BSE outbreak was greatly amplified by the recycling of the agent through the practice of feeding cattle meat-and-bone meal (MBM), containing rendered cattle carcasses, some of which died of BSE (Wells and Wilesmith, 2004). In the past, cattle feed was rendered using several treatment steps, including prolonged heating in the presence of a hydrocarbon solvent. In the late 1970s and early 1980s, the solvent-extraction steps were omitted, and this may have contributed to the emergence of BSE by allowing infectious prion concentrations to survive the rendering process. Other factors that may have contributed to the emergence of BSE in the United Kingdom include a relatively high rate of endemic scrapie, a high population ratio of sheep to cattle, and the inclusion of MBM at high rates in cattle feed.
In the UK, over 2 million cattle were estimated to have been infected with BSE. Approximately half of these cattle may have been slaughtered for human consumption, potentially exposing millions of UK residents. Beginning in 1988, several protective measures were implemented to prevent further exposure of animals and humans to BSE, including animal feed bans and removal of infectious cattle tissues such as the brain from human food. Infections of additional UK cattle with BSE dramatically declined after implementation of these protective measures.

BSE outside the United Kingdom was first reported in Ireland in 1989 and Portugal and Switzerland in 1990 (Table 3). By 2005, the number of countries reporting BSE among native cattle increased to 24; 20 of these were European countries. In most European countries, the BSE outbreak appears to be declining, although some cases continue to occur. The first North American BSE case was reported in 1993 in a cow imported into Canada from the UK. Rendered cohorts of this cow may have been responsible for the 14 BSE cases identified in Canada during 2003–07, including a BSE-positive cow identified in Washington State and traced to a Canadian farm. In the United States, BSE was confirmed in an approximately 12-year-old cow born and raised in Texas and a 10-yearold cow from Alabama. The source of BSE infection in the U.S. cows remains unknown but the Western blot characteristics of the prions seemed to be atypical (H-type), potentially representing a different BSE strain. Other BSE cases with unusual molecular phenotype have also been reported in other countries, including the L-type in Belgium, Italy, and Japan and the H-type in France and Germany.
Because cattle carcasses were included in the production of animal feed, potential BSE transmission to other animals was considered early during the BSE outbreak in the United Kingdom. BSE-like diseases were identified in zoo animals (ungulate spongiform encephalopathy) beginning in the late 1980s and in domestic cats (feline spongiform encephalopathy) beginning in 1990, indicating the potential for the BSE agent to cross the species barrier and spread to other animals. This development led to the reestablishment of CJD surveillance in the UK to monitor the possible transmission of BSE to humans.
Variant Creutzfeldt-Jakob Disease
In 1996, the UK National CJD Surveillance Unit reported a cluster of patients with a TSE having unusual but similar clinicopathologic manifestations (Will et al., 1996). Because the age and clinical and neuropathologic profile for the patients differed from those of classic CJD patients, the term variant CJD (vCJD) was used to describe this emerging TSE in humans. In subsequent years, several laboratory studies showed that the BSE and vCJD agents were indistinguishable, indicating that vCJD was a new disease and that its occurrence represented BSE transmission to humans. As of October 2007, 204 vCJD patients were reported worldwide, including 166 patients from the United Kingdom, 22 from France, 3 from Ireland, 2 each from the Netherlands, Portugal, and Saudi Arabia, and 1 each from Italy and Spain. The worldwide vCJD count includes 5 patients who likely acquired variant CJD during their past residence in the UK but were residents of Canada (1 patient), Ireland (1 patient), Japan (1 patient), and the United States (2 patients) at the time of illness onset.
Clinical and laboratory findings (Table 4) distinguish vCJD from the more common, classic CJD. Patients with vCJD have a younger median age at death (28 compared with 68 years), predominantly psychiatric manifestation at onset, delayed appearance of frank neurologic signs, pulvinar sign on MRI, virtual absence of typical EEG findings, and longer illness duration (14 compared with <6 months). All vCJD patients tested to date almost exclusively had methionine homozygosity at the polymorphic codon 129 of the prion protein gene. This homozygosity is present in approximately 35–40% of the general population. The neuropathology in vCJD is distinguished from that of classic CJD by the presence of numerous ‘florid plaques’ consisting of amyloid deposits surrounded by a halo of spongiform lesions.

In the UK, vCJD transmission has been reported in recipients of blood collected up to 3 years before vCJD onset in the donors (Llewelyn et al., 2004). Because many UK residents have potentially been exposed to BSE, concerns still exist about additional secondary spread of the agent via blood and possibly contaminated surgical instruments.
Human TSEs Associated With Genetic Mutations
One of the intriguing properties of human TSEs is that they can be both inherited and infectious. The inherited or genetic forms of TSEs are associated with insertion, deletion, or point mutations of the prion protein gene. At least 24 different point mutations have been described in association with human TSEs, having widely different clinical and neuropathologic manifestations (Kong et al., 2004). Historically, genetic forms of TSEs were classified as familial CJD, GSS, and FFI in part based on their phenotype.
A polymorphism at codon 129 of the human prion protein gene coding either for methionine or valine seems to markedly influence the clinicopathologic phenotype of human TSEs. The most striking example is the phenotype associated with codon 178 mutation which substitutes aspartic acid with asparagine. Patients who have this mutation in combination with methionine on the mutant allele at codon 129 present with the FFI phenotype whereas patients who have valine at codon 129 of the mutant allele present with the familial CJD phenotype.
Familial CJD
Familial CJD patients generally have clinicopathologic phenotype similar to nongenetic forms of CJD. Other family members are commonly affected with the disease because of a dominant inheritance pattern. Familial CJD has been reported among many family clusters from Canada, Europe, Japan, Israel, the United States, and several Latin American countries (Kong et al., 2004). It is most frequently associated with a mutation substituting glutamic acid with lysine at codon 200 of the prion protein gene. This mutation is the most common cause for inherited human TSEs. A majority of family members carrying the mutation eventually die of CJD. The largest familial cluster of codon 200 mutations was reported among Jews of Libyan and Tunisian origin. Fourteen other less frequent mutations associated with familial CJD have been reported from many countries.
Gerstmann-Straussler-Scheinker Syndrome
The term GSS is used to describe a heterogeneous group of inherited human TSEs characterized by a long illness duration (median about 5 years) and neuropathologic feature of numerous amyloid plaques primarily in the cerebellum. GSS is named after the three physicians who in 1936 reported the disorder in multiple generations of an Austrian family. The disease in this family was later shown to be associated with codon 102 mutation of the prion protein gene and may have been first identified in 1912. At least 13 different prion protein gene mutations in about 56 kindred have been reported in association with the GSS phenotype from Canada, Europe, Japan, Israel, Mexico, and the United States. GSS mutations are associated with a greater degree of variability in disease phenotype than other inherited TSEs. The most frequent GSS mutation results in leucine for proline substitution at codon 102 and is coupled with methionine at codon 129 of the mutant allele. Patients with this mutation commonly manifest with ataxia, dysarthria, movement disorders, and possibly dementia and akinetic mutism. The illness may last for 6 years in some patients with GSS 102 mutation. An illness duration exceeding 20 years has been reported in other forms of GSS.
Fatal Familial Insomnia
FFI patients have predominant involvement of the thalamus, resulting in severe sleep disturbances, often with intractable insomnia, and autonomic nervous system dysfunction, including abnormalities in temperature regulation, increased heart rate, and hypertension. Pathologic examination of the brain from FFI patients consistently shows preferential involvement of the thalamus with more severe neuronal loss and astrogliosis than seen in other brain regions. Although occasional sporadic fatal insomnia cases with no prion protein gene mutation have been identified, FFI is primarily associated with codon 178 mutation, resulting in the substitution of aspartic acid with asparagine in combination with methionine at codon 129 of the mutant allele. FFI patients have been reported from several European countries, Australia, Canada, Japan, and the United States. The clinical signs and illness duration in FFI patients are influenced by the polymorphism at codon 129 of the nonmutant allele.
Chronic Wasting Disease
CWD is a TSE that affects North American cervids in their natural habitat and captive environment. CWD’s known natural hosts are mule deer, white-tailed deer, elk, and moose. It was first identified as a fatal wasting syndrome of captive mule deer in the late 1960s in Colorado and Wyoming research facilities (Williams et al., 2002). It was recognized as a TSE in 1978 and among wild cervids in 1981. CWD can be highly transmissible in affected herds, in some cases reaching a prevalence of over 90% within several years. The mode of transmission among cervids is poorly understood. Transmission is believed to occur by direct animal-to-animal contact or indirect exposure to contaminated feed and water sources.
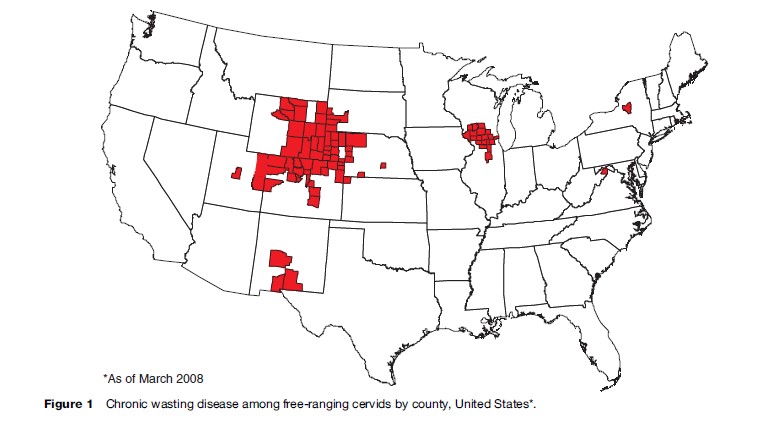
Surveillance and modeling studies have indicated that CWD may have occurred endemically for decades in a contiguous area in northeastern Colorado and southeastern Wyoming. Since 2000, CWD among free-ranging deer and elk has been increasingly identified in nine additional states and two Canadian provinces (Figure 1). CWD has been experimentally transmitted by intracerebral inoculation into other animals, including cattle, goats, squirrel monkeys, and laboratory mice. An in vitro cell-free experiment demonstrated inefficient conversion of human prion protein by CWD prions. However, no human TSEs with strong evidence of a link with CWD have been identified (Belay et al., 2004).
Bibliography:
- Belay ED (1999) Transmissible spongiform encephalopathies in humans. Annual Review of Microbiology 53: 283–314.
- Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, and Schonberger L (2004) Chronic wasting disease and potential transmission to humans. Emerging Infectious Diseases 10: 977–984.
- Kong Q, Surewicz WK, Peterson RB, et al. (2004) Inherited prion diseases. In: Prusiner SB (ed.) Prion Biology and Diseases, vol. 14, pp. 673–775Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
- Llewelyn CA, Hewitt PE, Knight RSG, et al. (2004) Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. The Lancet 363: 417–421.
- Parchi P, Giese A, Capellari S, et al. (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Annals of Neurology 46: 224–233.
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144.
- Wells GAH and Wilesmith JW (2004) Bovine spongiform encephalopathy and related diseases. In: Prusiner SB (ed.) Prion Biology and Diseases, vol. 12, pp. 595–628. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Will RG, Ironside JW, Zeidlem, et al. (1996) A new variant of CreutzfeldtJakob disease in the UK. The Lancet 347: 921–925.
- Will RG, Alper MP, Dormont D, and Schonberger LB (2004) Infectious and sporadic prion diseases. In: Prusiner SB (ed.) Prion Biology and Diseases, vol. 13, pp. 629–672. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Williams ES, Miller MW, Kreeger TJ, Khan RH, and Thorne ET (2002) Chronic wasting disease of deer and elk: A review with recommendations for management. Journal of Wildlife Management 66: 551–563.
- Belay ED and Schonberger LB (2005) The public health impact of prion diseases. Annual Review of Public Health 26: 191–212.
- Collee JG and Bradley R (1997) BSE: a decade on – Part 1. The Lancet 349(9052): 636–641.
- Collee JG and Bradley R (1997) BSE: a decade on – Part 2. The Lancet 349(9053): 715–721.
- Knight RS and Will RG (2004) Prion disease. Journal of Neurology, Neurosurgery, and Psychiatry 75(Suppl 1): i36–i42.
- Prusiner SB (ed.) (2004) Prion Biology and Diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- http://www.aphis.usda.gov/newsroom/hot_issues/bse/index.shtml – Animal and Plant Health Inspection Service, U.S. Department of Agriculture.
- http://www.defra.gov.uk/animalh/bse/index.html – Bovine Spongiform Encephalopathy (BSE), Department for Environment, Food, and Rural Affairs, Animal Health and Welfare (UK).
- http://www.bseinquiry.gov.uk – The BSE Inquiry, Inquiry into BSE and Variant CJD in the UK.
- http://www.cjdfoundation.org – Creutzfeldt-Jakob Disease (CJD) Foundation.
- http://www.cjdsurveillance.com – National Prion Disease Pathology Surveillance Center.
- http://www.cdc.gov/ncidod/dvrd/prions/index.htm – Prion Diseases, Centers for Disease Control and Prevention (CDC).
See also:
Free research papers are not written to satisfy your specific instructions. You can use our professional writing services to buy a custom research paper on any topic and get your high quality paper at affordable price.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality



